

MPS III (Sanfilippo A,B,C et D)
MPS III A et MPS III B ; point sur les essais de thérapie génique en cours
Pour faire suite aux présentations orales faites à l’occasion du congrès WORLD 2020 (We’re Organizing Research on Lysosomal Deseases) à Orlando, nous vous proposons un retour sur des résultats intermédiaires pour trois essais cliniques sur les maladies de Sanfilippo types A et B. Si vous souhaitez plus de précisions, n’hésitez pas à contacter l’association.
Résultats intermédiaires de l’essai clinique Transpher A, de thérapie génique par ABO-102 dans la MPS III A (laboratoire Abeona)
Pour rappel, l’étude Transpher A est un essai clinique de phase I-II pour la maladie de Sanfilippo A. Il s’agit d’une thérapie génique par administration intraveineuse d’un vecteur AAV9 contenant le gène hSGSH impliqué dans cette maladie.
Les objectifs premiers de cette étude sont d’évaluer la sécurité de ce traitement et l’évolution du développement neurocognitif des patients traités en la comparant, à âge équivalent, à celle observée chez des enfants malades non traités (histoire naturelle de la maladie). D’autres critères dits secondaires sont également évalués (volume du foie, de la rate, du cerveau par IRM ; tests de comportement, questionnaires de qualité de vie…).
Trois groupes successifs de malades ont reçu, en une injection unique, le vecteur en quantité différente (escalade de dose). L’augmentation de dose n’était faite que si aucun événement négatif important ne survenait dans le groupe traité à la dose inférieure. Le critère de l’âge, imposé pour l’inclusion, a été modifié durant l’étude et est désormais compris entre 6 mois et 2 ans. Si l’enfant a plus de 2 ans, il doit avoir un quotient de développement égale ou supérieur à 60 (utilisation de l’échelle de Bayley). Le suivi des enfants traités se fait sur une période initiale de deux ans, suivie d’une période d’extension de trois ans supplémentaire.
Trois patients constituent le premier groupe (faible dose) avec un suivi moyen de 43 mois. Le second groupe comporte également 3 patients traités à moyenne dose dont le suivi moyen est de 35 mois. Enfin le troisième groupe, pour ces analyses intermédiaires, était constitué de 8 malades (inclusion de nouveaux malades toujours en cours) traités à forte dose pour un suivi moyen de 23 mois.
Dans l’ensemble le traitement est bien toléré. On observe une rapide diminution de l’héparane sulfate (substance en surcharge dans cette maladie) dans le liquide céphalo-rachidien (liquide « baignant » la moelle et le cerveau), diminution d’autant plus marquée que la dose de vecteurs injectés est importante. Une diminution importante transitoire du l’héparane sulfate plasmatique, la remontée reste néanmoins inférieure au niveau de départ. Le volume du foie est également fortement et durablement diminué. Les trois plus jeunes enfants traités (à la plus forte dose) continuent à progresser sur le plan neurocognitif mais le recul est encore insuffisant pour confirmer véritablement une évolution différente de celle de l’Histoire Naturelle de la maladie.
Point sur l’essai clinique AAVance, thérapie génique par AAVrh10 dans la MPS III A (laboratoire Lysogène)
Il s’agit d’un essai clinique de phase II/III dont l’objectif premier est de démontrer l’efficacité du traitement par un ralentissement du déclin du neurodéveloppement de chaque enfant malade à 2 ans post traitement. D’autres critères secondaires sont évalués durant cette étude : la sécurité et la tolérance au traitement, l’effet sur le comportement, sur le sommeil, sur la qualité de vie.... Il est prévu d’inclure 20 patients (la fin des inclusions est prévue au premier semestre 2020) qui seront suivis durant 2 ans puis 3 ans supplémentaires dans une phase dite d’extension. Le premier patient a été traité en février 2019 par une injection directe dans le cerveau (ICV) du vecteur AAVrh10 contenant le gène SGSH impliqué dans la maladie de Sanfilippo A. Parmi les critères d’inclusion, les malades doivent être âgés d’au moins 6 mois avec un quotient de développement (QD) égale ou supérieur à 50 (un QD = 100 correspond à la moyenne obtenue sur le test chez des enfants du même âge non malade).
Préalablement à cet essai, le laboratoire a mené, chez 23 patients, une étude prospective sur 2 ans, de l’histoire naturelle de la maladie. Cette étude montre que ces enfants atteignent leur performance maximale au plus tard à 4,5 ans, aucun ne dépassant un quotient de développement équivalent à celui d’un enfant non malade de 35 mois. L’étude montre également que la plus grande variation de QD, d’un enfant à l’autre, se trouve entre 3 et 6 ans, certains pouvant suivre une courbe de QD identique aux enfants non malades jusqu’à 2-3 ans. A partir de cette étude, un modèle mathématique prédictif du déclin a été obtenu pour servir de référence à l’essai clinique AAVance. En parallèle, le laboratoire développe une nouvelle exploration de critères d’évaluation (communication, écoute, hyperactivité, jeux en interactions, marche…) à partir de vidéos obtenues en vie réelle chez les enfants impliquées dans l’étude AAVance.
En décembre 2019, 15 enfants étaient traités avec un temps de suivi compris entre 1 et 11 mois.
Point sur l’essai clinique Transpher B, thérapie génique par ABO-101 dans la MPS III B (laboratoire Abeona)
Cette étude est similaire à Transpher A. Il s’agit d’un essai clinique de phase I/II d’un traitement par une injection intraveineuse d’un vecteur AAV9 contenant le gène humain NAGLU impliqué dans la maladie de Sanfilippo B. Le but premier est d’en évaluer la sécurité et de comparer l’évolution du développement neurocognitif des patients traités à celle d’enfants non traités du même âge (histoire naturelle). Les patients inclus sont âgés entre 6 mois et 2 ans, ou si plus de 2 ans ont un QD égal ou supérieur à 60.
Un premier groupe constitué de 2 patients ont reçu le traitement à faible dose (13 et 26 mois de suivi post traitement), puis un second groupe de 5 patients ont reçu le traitement à plus forte dose (entre 2,3 et 9 mois de suivi). Dernièrement la constitution d’un troisième groupe d’enfants traités à une dose plus importante a été amendé au protocole de l’étude. Le premier enfant de ce groupe a été traité en janvier 2020.
Jusqu’à la date de la présente étude, le traitement est bien toléré. La période de suivi des marqueurs est courte mais montre pour l’instant une diminution du taux de l’héparane sulfate dans le liquide céphalo-rachidien, le plasma et les urines ainsi qu’une réduction des glycosaminoglycanes (GAGs) dans les urines et une diminution de la taille du foie. Le recul est insuffisant pour avoir des informations sur l’évolution du développement neurocognitif.
[/Delphine GENEVAZ/ Mai 2020]
Présentation par le laboratoire Lysogene des principaux résultats de l’essai clinique de thérapie génique chez les enfants MPS IIIA
Mi-novembre, le laboratoire Lysogene, dans un communiqué de presse suivi d’un webcast, informait sur les principaux résultats de son essai clinique de thérapie génique dans la maladie de Sanfilippo A.
Pour rappel, dix neufs enfants, âgés entre 10 et 64 mois, ont été inclus dans cet essai clinique de phase 2/3. Ils devaient avoir un quotient de développement (QD) égale ou supérieur à 50 (un QD de 100 correspond à la moyenne obtenue sur le test par des enfants du même âge non malades). Les enfants participant à l’essai ont reçu, par une injection directe dans le cerveau, un vecteur contenant le gène impliqué dans la maladie de Sanfilippo A.
L’analyse des résultats s’est faite sur deux cohortes, la principale constituée des 13 enfants âgés de 30 mois et plus, et la cohorte secondaire, dite ancillaire, composée par les 6 enfants âgés de moins de 30 mois.
Pour la cohorte principale, l’étude n’a pas atteint son critère premier d’efficacité, à savoir une amélioration statistiquement significative du QD 24 mois après le traitement, en comparaison aux valeurs du QD mesurées chez des enfants non traitées lors d’une étude de l’histoire naturelle effectuée précédemment. Les autres critères secondaires d’efficacité n’ont également pas été atteints : stabilisation ou amélioration de l’âge du développement cognitif, du développement associé au langage, du développement associé aux fonctions motrices, à 24 mois post-traitement en comparaison aux valeurs obtenues lors de l’inclusion.
Pour la seconde cohorte, le critère principal était atteint avec une amélioration statistiquement significative du développement cognitif à 24 mois post traitement en comparaison avec les patients ayant participé à l’histoire naturelle. Le déclin moyen (en pourcentage) du QD sur 24 mois était inférieur de 27 % à celui des patients de l’histoire naturelle au même âge, ce ralentissement du déclin étant variable individuellement. Dans cette cohorte, les patients ayant atteint l’âge de 40 mois plus ou moins 3 mois (5 enfants sur 6), l’âge de développement cognitif moyen était 48% plus élevé que ceux des patients du même âge de l’histoire naturelle. Il a été également démontré une stabilisation ou amélioration statistiquement significative de l’âge du développement du langage et moteur (critères d’efficacité secondaire) et pas de diminution statistiquement significative des scores aux tests évaluant le comportement adaptatif global, la communication, les aptitudes à la vie quotidienne, la socialisation et les aptitudes motrices, entre les valeurs initiales et 24 mois après le traitement. De même aucune diminution significative du volume de la matière grise corticale n’a été observée suggérant une préservation à 24 mois post traitement de la fonction cognitive pour cette cohorte. Une analyse complète sera faite au second trimestre 2023, lorsque l’ensemble des patients inclus avant l’âge de 30 mois auront plus de 48 mois.
Concernant la tolérance et l’innocuité du traitement, des anomalies dans la substance blanche au niveau des points d’injection ont été observées. Ces lésions se sont stabilisées ou ont diminuées chez la plupart des enfants, n’entrainant l’observation d’aucun symptôme clinique significatif.
Le laboratoire indique que ces résultats permettent de mieux préciser la population de patients qui pourrait bénéficier du traitement.
Vous pouvez retrouver la présentation des résultats lors du webcast avec le lien suivant : LYSOGENE
Comprendre
Décrite en 1963 par le pédiatre américain Sylvester Sanfilippo, la maladie de Sanfilippo (ou sous son terme scientifique : mucopolysaccharidose de type III (MPS III)) est une affection génétique rare qui fait partie des maladies de surcharge lysosomale.
Elle est due à une accumulation anormale de molécules de type sucré (sucre = saccharose >> maladie de type mucopolysaccharidose) dans les cellules de l’organisme, plus particulièrement celles du cerveau.
La maladie de Sanfilippo est divisée en quatre sous-types qui présentent tous des symptômes similaires. En revanche sur le plan biochimique ils sont différents. Chaque sous-type est déterminé en fonction de l’enzyme qui est absente ou déficitaire.
On distingue ainsi :
- la mucopolysaccharide de type III A (Sanfilippo A)
- la mucopolysaccharide de type III B (Sanfilippo B)
- la mucopolysaccharide de type III C (Sanfilippo C)
- la mucopolysaccharide de type III D (Sanfilippo D)
Quelles enzymes interviennent dans la maladie de Sanfilippo ?
Il y a quatre enzymes qui interviennent dans cette maladie. L’absence de l’une d’entre elles est responsable d’un sous-type de la maladie :
- l’héparane-N-sulfatase ou héparane sulfamidase est déficitaire dans la MPS III A
- l’alpha-N-acétylglucosaminidase est déficitaire dans la MPS III B
- l’acétyl-CoA:alpha-glucosaminide-N-acétyltransférase est déficitaire dans la MPS III C
- N-acétylglucosamine-6-sulfatase est déficitaire dans la MPS III D
Lorsqu’une de ces quatre enzymes est absente, cela provoque une accumulation d’héparane sulfate dans les lysosomes.
Ce dysfonctionnement est dû à des mutations dans le gène correspondant à l’enzyme en question. Ces mutations génétiques font que l’enzyme n’est pas ou peu fabriquée. Ces quatre enzymes interviennent à différents niveaux de dégradation mais si l’une d’elles est absente ou défectueuse, les conséquences sont les mêmes.
Comment se transmet la maladie de Sanfilippo ?
La transmission de cette maladie se fait sur un mode autosomique récessif. Les deux parents sont "porteurs sains" et transmettent leurs deux gènes mutés à l’enfant.
Combien de personnes sont atteintes de la maladie ?
Même si elle reste rare, la maladie de Sanfilippo est la plus fréquente des mucopolysaccharidoses (MPS). En France, elle concerne aujourd’hui moins d’une naissance sur 100 000. Le nombre de nouveaux cas observés dans la population diffère toutefois selon le sous-type.
Le nombre de patients n’est pas connu avec justesse, il est estimé entre 100 et 150 malades en vie dont prés de 65% sont MPS IIIA, 24%MPS IIIB, 9% MPS IIIC et quelques rares MPSIIID.
Les estimations se basent entre autre sur un travail d’Histoire naturelle qui a été effectué entre 1990 et 2006, et des études entre 2000 et 2013. VML collabore actuellement à la création d’un registre national MPS afin de mieux quantifier et connaitre l’évolution de la maladie.
Ces sous-types sont par ailleurs plus fréquents dans certaines populations. Par exemple, la MPS III A est plus fréquente en Europe du Nord alors qu’elle est absente en Grèce, où c’est la MPS III B qui prédomine.
Quels sont les symptômes ?
Généralement, les premiers symptômes surviennent avant 6 ans.
Dans un premier temps, ce sont les troubles du comportement et les problèmes de concentration qui vont interpeller les parents. Hyperactivité, irritabilité, agressivité et anxiété sont par exemple souvent observées. L’enfant peut aussi avoir un retard du développement du langage. Dans ce cas, quelques difficultés à articuler et à acquérir le langage c’est-à-dire la capacité à associer deux mots, sont rencontrées. Par ailleurs, certains enfants présentent des troubles du sommeil qui peuvent être parfois importants. D’autres, au contraire, ne sont absolument pas concernés par ce symptôme. Les infections du nez, des oreilles et de la gorge sont quant à elles fréquentes chez la majorité des patients. Ces premiers symptômes évoluent ensuite progressivement et plus ou moins rapidement en fonction des individus.
Les symptômes et signes physiques sont peu visibles dans les premières années, mais vont progressivement évoluer. On pourra notamment noter chez les patients des traits du visage un peu plus épais et une implantation épaisse de cheveux et sourcils.
Dans un second temps, les enfants sont généralement plus calmes. Ils peuvent toutefois développer pour certains une épilepsie. Les troubles du sommeil peuvent être quant à eux toujours présents. Les patients sont alors dans une phase relativement stable et plus ou moins longue selon les personnes et le sous-type de la maladie.
Dans un troisième temps, un net avancement de l’atteinte neurologique est observé. Les acquisitions psychomotrices diminuent progressivement avec la perte de la marche et de la station assise. Des complications orthopédiques peuvent aussi aggraver la difficulté de déplacement des patients. Il peut y avoir par ailleurs un affaiblissement de la vue et de l’ouïe ainsi que des troubles de l’alimentation et du transit. La communication orale est de plus en plus affectée.
Souvent dû à des complications respiratoires, le décès peut arriver vers la fin de la deuxième décade mais certaines personnes vivent plus de 50 ans. L’âge du décès est très variable suivant les individus et le sous-type de mucopolysaccharidose de type III.
Quels sont les traitements ?
Jusqu’à ce jour, aucun traitement spécifique n’a été mis au point mais plusieurs sont actuellement à l’étude. Des essais cliniques de thérapie génique sont en cours pour la maladie de Sanfilippo A et Sanfilippo B. Sinon seuls des traitements symptomatiques sont aujourd’hui disponibles et peuvent avoir un effet bénéfique sur le comportement, le sommeil, des douleurs neuropathiques, etc ...
Diagnostic
D’après les témoignages des parents d’enfants malades, c’est lorsqu’ils constatent un retard de langage durant les années préscolaires de leur enfant, qu’ils commencent à s’inquiéter. Ce retard n’est pas uniquement constaté dans la maladie de Sanfilippo mais il fait partie des principaux symptômes observés. Le langage, s’il a été acquis, et la compréhension se dégradent également peu à peu et plus ou moins lentement selon les personnes.
C’est l’observation d’un ensemble de symptômes qui doit conduire le médecin à envisager une maladie de Sanfilippo. Le diagnostic de la maladie de Sanfilippo ne pourra être posé qu’après une mesure de la concentration d’héparane sulfate dans les urines. En cas de maladie, une concentration d’héparane sulfate plus importante est détectée.
Ce diagnostic est ensuite confirmé par un dosage enzymatique dans les leucocytes ou les fibroblastes pour les quatre sous-types ou dans le sérum pour la MPS III B. Cela permet de constater un déficit ou une absence d’activité de l’une des quatre enzymes. Cette étape est indispensable pour confirmer le diagnostic et préciser le sous-type.
Toutefois, pour les MPS III A et D, la mesure de l’activité d’autres sulfatases est essentielle pour exclure une autre maladie lysosomale, la maladie d’Austin, qui est justement due à un déficit en sulfatases.
Le sous-type étant suggéré, ce sont des analyses génétiques qui vont, à ce stade, confirmer la maladie. Une recherche de mutations sur le gène codant l’enzyme en question est effectuée.
Pour obtenir une détection fiable des "porteurs sains" dans les familles, l’identification des mutations chez le malade est importante. Plus de 70 mutations sur le gène codant l’héparane sulfamidase ont aujourd’hui été identifiées.
Un diagnostic précoce suivi de soins appropriés joue un rôle important sur la qualité de vie des patients. Cela peut même parfois ralentir ou prévenir le développement de dommages irréversibles.
Toutes les étapes du diagnostic sont importantes pour être certain que la personne soit bien atteinte de la maladie de Sanfilippo.
Existe-t-il un dépistage de la maladie de Sanfilippo ?
Un test génétique est en effet possible. Il sera proposé aux couples ayant déjà un enfant malade (ou famille à risque dû à des antécédents), permettant ainsi d’envisager une grossesse en toute sécurité. Le test peut aussi permettre de diagnostiquer précocement la fratrie d’un enfant malade.
>> Pour les couples ayant déjà eu un enfant atteint (ainsi que les couples où chacun a été dépisté comme porteur sain et les familles à risque dû à des antécédents) peuvent bénéficier du diagnostic prénatal. Il est actuellement réalisé par un test génétique.
Il est donc indispensable d’avoir au préalable identifié les mutations en cause chez le(s) malade(s), et vérifié que les deux parents demandeurs du diagnostic prénatal soient chacun porteur d’une des mutations familiales (qui peuvent être différentes ou identiques). Il est donc souhaitable de réaliser ces études dès le diagnostic du malade. Dans ces conditions, le résultat est fiable, sous réserve que l’on ait pu vérifier que les cellules qui ont servi à extraire l’ADN utilisé pour le test sont bien celles du fœtus et non celles de la mère (test complémentaire indispensable).
>> Pour la fratrie, il est possible de dépister cette maladie chez les personnes à risque (frères et sœurs du malade) avant qu’elle ne se déclare. Les tests génétiques permettent d’identifier dans la famille du malade les porteurs sains et les personnes atteintes de la maladie qui n’auraient pas encore développé les symptômes. En cas de résultats positifs, ce dépistage et diagnostic précoces vont permettre une meilleure prise en charge et limitation des symptômes mais en l’absence de traitement spécifique, ils n’assurent pas une guérison.
Sur le plan génétique, qui est atteint de la maladie de Sanfilippo ?
La maladie de Sanfilippo est une maladie génétique dite autosomique récessive.
Notre matériel génétique fonctionne par paire. Le malade possède deux copies défectueuses sur l’un des quatre gènes impliqués dans la maladie de Sanfilippo. Ces deux gènes défectueux lui ont été transmis par ses parents, qui possèdent eux un seul gène défectueux, c’est pourquoi ils sont dits "porteurs sains". Dans ce cas, le gène muté du père doit être le même que le gène muté de la mère. Par exemple, si les deux parents transmettent chacun une copie altérée du gène impliqué dans la MPS III B, alors leur enfant sera atteint de la MPS III B.
Au contraire, si les parents portent chacun une mutation différente, il n’y a aucun risque pour les enfants de développer la maladie de Sanfilippo. En effet, le gène muté du père (sur un seul chromosome de la paire) est différent de celui de la mère. Les enfants peuvent être porteurs sains pour les deux sous-types en question ou pour l’un ou l’autre des deux sous-types. Par exemple, si le père transmet une copie altérée du gène impliqué dans la MPS III A et si la mère transmet une copie altérée du gène impliqué dans la MPS III D, leur enfant sera porteur sain pour ces deux sous-types et ne développera donc ni la MPS III A ni la MPS III D.
Traitements
Des essais cliniques de thérapie génique sont en cours pour la maladie de Sanfilippo A et Sanfilippo B.
Sinon pour l’instant, seuls des traitements symptomatiques sont disponibles. Ils sont indispensables pour essayer d’améliorer la qualité de vie du malade et de ses proches.
Des médicaments sont ainsi utilisés pour traiter les divers troubles : problèmes neurologiques (antidépresseurs, antiépileptiques, antipsychotiques, myorelaxant…), troubles du sommeil, etc.
Dans quelques cas, une dérivation du liquide céphalo-rachidien a permis d’améliorer des troubles du comportement réfractaires au traitement pharmacologique.
Une prise en charge non médicamenteuse est également possible. Souvent mal acceptés, des appareillages orthopédiques et des soins de kinésithérapie pour entretenir les articulations sont nécessaires. Ils permettent en effet de préserver la mobilité de l’enfant le plus longtemps possible. Par ailleurs, si l’enfant a des problèmes auditifs, il peut y avoir un certain temps d’adaptation pour qu’il tolère l’appareil. D’autre part, s’il a besoin de soins dentaires, une anesthésie lui est généralement faite. Cela permet en effet de prendre toutes les précautions pour garantir le bon déroulement de la séance. D’une manière générale, la personne doit être régulièrement vue par le médecin qui adaptera le traitement en fonction de l’état du patient.
Il a été observé par expérience que la stimulation régulière et continue de l’enfant est aussi un moyen de ralentir les pertes d’acquisition.
Plusieurs traitements spécifiques sont actuellement à l’étude comme :
- la thérapie de réduction du substrat de l’enzyme déficiente. Elle correspond à l’utilisation de Génistéine qui a plusieurs activités biologiques qui pourraient être impliquées dans l’effet positif observé lors de son utilisation dans les études précliniques.
- la transplantation de cellules souches hématopoïétiques.
- la thérapie génique. Des essais cliniques pour la MPS III A et la MPS III B sont actuellement menés. Ils consistent à injecter dans le cerveau, par neurochirurgie, une certaine concentration d’un vecteur contenant le gène correct de l’enzyme impliquée dans le sous-type en question. L’objectif premier est d’évaluer la tolérance et l’innocuité de ce traitement.
- la thérapeutique enzymatique substitutive par voie intrathécale. Elle correspond à l’injection de l’enzyme déficiente au niveau de la colonne vertébrale dans le liquide céphalo-rachidien via un dispositif mis en place de type port-à-cath (boîtier implanté sous la peau). Des essais cliniques pour la MPS III A sont actuellement en cours pour vérifier l’innocuité de ce type de traitement.
La thérapeutique enzymatique substitutive par voie intraveineuse n’est pas disponible car l’enzyme manquante ou déficiente ne peut pas traverser la barrière hémato-méningée qui est un filtre extrêmement sélectif qui protège normalement le cerveau des agents pathogènes et des toxines du sang.
Quand ces traitements doivent-ils débuter ?
Pour être totalement efficaces, ces traitements doivent être commencés avant qu’il y ait des dommages irréversibles du système nerveux central (troubles neurologiques). Si un retard de développement est observé, le système nerveux central est déjà touché. Un diagnostic précoce est donc indispensable.
VML, votre association
VML est une association POUR et PAR les patients, parents, familles touchés par les maladies lysosomales... Chacun pourra y trouver information, conseil, partage, accompagnement, mais pourra aussi apporter à son tour son envie d’agir contre la maladie et le handicap pour qu’ensemble nous fassions avancer le combat !
>> Retrouvez d’autres parents et patients au sein du Groupe VML-Sanfilippo.
Si l’une des forces de VML est de fédérer l’ensemble des maladies lysosomales, les parents et patients concernés par la maladie de Sanfilippo peuvent aussi se retrouver plus spécifiquement dans le "groupe patho VML-Sanfilippo". Réunis autour d’un référent volontaire, les participants ont pour habitude de partager et d’échanger par mail leurs expériences, leurs informations, etc. Le groupe peut aussi être porteur d’un projet spécifique et son activité est liée à l’action des membres du groupe. Chacun peut être actif et intervenir à tout moment de l’année. Les membres se rencontrent généralement à l’occasion du week-end annuel de VML qui a lieu en mai, ou lors d’une journée spécifique Sanfilippo.
>> Le livret génétique et transmission dans la maladie de Sanfilippo
Un livret VML sur la maladie de Sanfilippo est disponible sur simple demande à l’association. Il regroupe tout ce qu’il faut savoir sur la maladie (symptômes, traitements, diagnostic, transmission...).
>> Des fiches d’information
Parce qu’un certain nombre de questions reviennent fréquemment sur le plan social, VML édite des fiches d’informations pour aider ses adhérents dans leur quotidien (sécurité sociale et complémentaire, la PCH en établissement, droits MDPH, passage enfant-adulte...).
>> Des professionnels à vos côtés
Les adhérents de l’association VML peuvent s’appuyer sur une équipe professionnelle pour obtenir des informations, des réponses à leurs questions, de l’accompagnement et du soutien dans leur démarche. Ce service est assuré par la responsable scientifique, la psychologue et l’assistante sociale de l’association VML.
>> Les actions et missions de l’association VML
Consultez un aperçu de nos actions au profit des adhérents de l’association
>> Accompagner l’après
Ce n’est pas dans l’ordre des choses que de voir partir son enfant, de jeunes adultes ou son conjoint. Dans ces moments-là, l’association VML reste à vos côtés et propose des temps de rencontres spécifiques.
Témoignages
Retrouvez sur dailymotion :
- le reportage sur les Hôpitaux d’excellence
- l’interview d’un médecin sur les maladies lysosomales
- le reportage sur un essai clinique de la maladie de Sanfilippo en 2008 (témoignages de familles et de médecins). Les résultats de cet essai n’ont pas été concluants mais ont permis de faire avancer la recherche d’un traitement.
Professionnel
Mise en garde : Les informations figurant dans cette fiche détaillée sont rédigées par un ou plusieurs experts. Elles sont destinées aux professionnels de la santé. Il est donc important d’analyser ces informations avec son médecin traitant, à la lumière de sa propre situation.
La maladie de Sanfilippo ou mucopolysaccharidose de type III (MPS III) est une maladie de surcharge lysosomale, du groupe des mucopolysaccharidoses, caractérisée par une dégradation intellectuelle sévère et rapide. Cette maladie est constituée de quatre sous-types cliniquement proches mais biochimiquement distincts.
La maladie est sous-diagnostiquée (dysmorphie généralement peu marquée) ; c’est la plus fréquente des mucopolysaccharidoses en Hollande et en Australie avec une prévalence respective de 1/53 0000 et 1/67 000. La fréquence des différents sous-types varie selon les pays : sous-type A plus fréquent en Angleterre, Hollande et Australie, et sous type B plus fréquent en Grèce et Portugal, les types IIIC et IIID étant beaucoup plus rares.
Source : Atlas Médical VML- Docteur Bénédicte Héron, Professeur Irène Maire - Docteur Roseline Froissart - Février 2007
Métabolisme - Pathogénie - Génétique
Les enzymes déficitaires sont :
- l’héparane-N-sulfatase ou héparane sulfamidase pour la MPS IIIA,
- l’alpha-N-acétylglucosaminidase pour la MPS IIIB,
- l’acétyl-CoA :alpha-glucosaminide-N-acétyltransférase pour la MPS IIIC,
- la N-acétylglucosamine-6-sulfatase pour la MPS IIID.
Ces quatre enzymes sont indispensables pour la dégradation de l’héparane sulfate. Le déficit enzymatique entraîne donc une accumulation d’héparane sulfate dans les lysosomes de l’organisme, notamment du cerveau et son excrétion anormale dans les urines où il peut être détecté.
Les gènes de trois de ces quatre enzymes ont été localisés et clonés (celui de la MPS IIIA en 17q25.3 ; celui de la MPS IIIB en 17q21.1 ; celui de la MPS IIID en 12q14). Celui de la MPS IIIC a été localisé récemment sur le chromosome 8 en 8p11-8 q11. La transmission des quatre maladies de Sanfilippo se fait sur un mode récessif autosomique.
Les héparane sulfates jouent en particulier un rôle de régulateur dans la migration cellulaire, la guidance axonale, la synaptogénèse et la plasticité structurale des cellules neuronales expliquant que leur présence excessive puisse induire des dysfonctionnements importants au niveau cérébral. En outre, des changements secondaires dans les fonctions métaboliques et cellulaires ont été mis en évidence dans des modèles de MPS III (augmentations secondaires d’autres hydrolases acides, augmentation des concentrations en gangliosides GM2 et GM3, phénomènes inflammatoires,…). La relation entre ces modifications et les désordres neurologiques n’est pas élucidée.
Presentation Clinique

- Arnaud F. à 2 ans
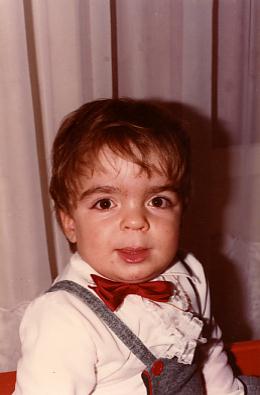

- Arnaud F. à 4 ans
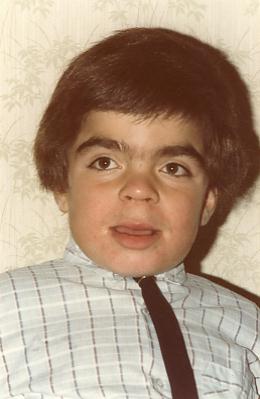
Signes de début
Les premiers symptômes de cette maladie apparaissent habituellement entre 2 et 6 ans. Il s’agit principalement de troubles du comportement (hyperkinésie, agressivité) associés à des troubles du sommeil parfois importants, et à une stagnation des acquisitions (retard de langage, contrôle sphinctérien rarement acquis) et alors que les signes dysmorphiques sont modérés. Dans les formes précoces, des accès de terreur inexpliqués peuvent survenir dès les premiers mois de vie : le nourrisson hurle et déclenche un pseudo-Moro comme s’il tombait dans le vide ; l’enfant plus grand peut se mettre à pleurer en boucle comme s’il avait mal ou était inconfortable.
Ces enfants ont souvent une macrocéphalie, une taille normale voire une avance staturale au début, des traits un peu grossiers, un hirsutisme avec des cheveux épais et drus. Une hépatosplénomégalie modérée peut être retrouvée surtout chez les enfants jeunes. Des otites à répétition sont fréquemment notées depuis la première année de vie, avec une hypertrophie éventuelle des végétations adénoïdes et des amygdales, et une atteinte auditive mixte fréquente.
Les signes ostéo-articulaires sont plus discrets ou tardifs : un discret flessum des coudes est fréquent ; la raideur d’autres articulations (épaules, hanches, genoux) est plus tardive. Sur le plan radiologique, un éventuel aspect de rostre (sur les dernières vertèbres dorsales ou la première vertèbre lombaire) ou de dysostose multiple (habituellement minime ici) sont des éléments d’orientation aspécifiques vers une MPS.
La découverte d’un souffle cardiaque peut correspondre à une dysplasie valvulaire mitrale ou aortique, mais l’atteinte de la fonction cardiaque est tardive. Les épisodes récurrents de diarrhée motrice ont tendance à s’estomper avec l’âge. La survenue d’une puberté précoce est possible.
Evolution
Une déformation cypho-scoliotique du rachis ou une ostéochondrite des têtes fémorales complique plus rarement l’évolution orthopédique que dans les autres mucopolysaccharidoses.
L’évolution est marquée par une progression nette de l’atteinte neurologique à partir de la fin de la première décennie : régression puis perte des acquisitions psychomotrices (apraxie puis perte de la marche, de la station assise, de l’intérêt pour l’environnement et de la mastication-déglutition), atteinte pyramidale et/ou manifestations extrapyramidales (akinésie, rigidité plastique ou tremblements d’action ; parfois syndrome dystonique, ou dyskinétique souvent accentué ou favorisé par un traitement neuroleptique).
Une rétinopathie pigmentaire peut être observée. Une neuropathie périphérique, se manifestant par une amyotrophie et des troubles vasomoteurs à prédominance distale (en gants et en chaussettes) avec hyporéflexivité ostéotendineuse , peut être source de douleurs ; elle est de type axonal sensitivo-moteur à l’électromyogramme.
Une épilepsie survient souvent au cours de l’évolution, chez le grand enfant ou l’adolescent : les crises épileptiques sont le plus souvent généralisées et peu fréquentes, bien contrôlées par une monothérapie anti-épileptique.
L’imagerie cérébrale (scanner ou IRM) montre une atrophie corticale puis cortico-sous-corticale progressivement sévère tandis que se complètent la perte d’autonomie sociale, motrice, alimentaire et la perte de communication.
Les enfants atteints de cette maladie décèdent habituellement à la fin de la deuxième décennie dans un tableau de dégradation psychomotrice très sévère, associée à une tétraplégie spastique avec ses complications orthopédiques, des troubles de l’alimentation et du transit, des complications respiratoires qui nécessitent un traitement symptomatique aussi précoce et adapté que possible.
Les phénotypes cliniques des maladies de Sanfilippo A, B, C et D sont assez semblables mais sont hétérogènes y compris parfois dans une même famille. Le type A est plus souvent sévère avec un début plus précoce, et un décès vers la fin de la deuxième décade, mais des patients adultes ont été rapportés dont un cas de cardiomyopathie isolée et révélatrice chez un patient adulte. Les types B, C et D sont plus hétérogènes.
Diagnostic biologique
La mise en évidence de cellules de surcharge (cellules de Gasser,…) dans le sang ou la moelle peut constituer un élément d’orientation mais est peu spécifique.
Le diagnostic biochimique
![]() L’étude des glycosaminoglycanes (mucopolysaccharides) dans l’urine permet de mettre en évidence une présence anormale et isolée d’héparane sulfate confortant l’orientation diagnostique vers une maladie de Sanfilippo. Mais cette recherche peut être négative si l’on se contente d’une mesure quantitative ne séparant pas les différentes fractions excrétées de mucopolysaccharides, en particulier dans les formes modérées de l’adulte, car l’excrétion urinaire d’héparane sulfate diminue au-delà de l’adolescence. L’étude des glycosaminoglycanes urinaires s’est révélée complètement négative à la fois au plan quantitatif et qualitatif, dans une famille d’adultes atteints de MPS IIID à forme extrêmement modérée rapportée dans la littérature.
L’étude des glycosaminoglycanes (mucopolysaccharides) dans l’urine permet de mettre en évidence une présence anormale et isolée d’héparane sulfate confortant l’orientation diagnostique vers une maladie de Sanfilippo. Mais cette recherche peut être négative si l’on se contente d’une mesure quantitative ne séparant pas les différentes fractions excrétées de mucopolysaccharides, en particulier dans les formes modérées de l’adulte, car l’excrétion urinaire d’héparane sulfate diminue au-delà de l’adolescence. L’étude des glycosaminoglycanes urinaires s’est révélée complètement négative à la fois au plan quantitatif et qualitatif, dans une famille d’adultes atteints de MPS IIID à forme extrêmement modérée rapportée dans la littérature.
![]() La mise en évidence du déficit enzymatique se fait par la mesure de l’activité des différentes enzymes dans le sérum (MPS IIIB seulement) ou les leucocytes et les fibroblastes (pour les quatre sous-types). Pour les types IIIA et IIID, la mesure d’une autre sulfatase est indispensable pour exclure un déficit multiple en sulfatases (maladie d’Austin). L’identification des mutations chez le malade permet une détection fiable des hétérozygotes dans les familles.
La mise en évidence du déficit enzymatique se fait par la mesure de l’activité des différentes enzymes dans le sérum (MPS IIIB seulement) ou les leucocytes et les fibroblastes (pour les quatre sous-types). Pour les types IIIA et IIID, la mesure d’une autre sulfatase est indispensable pour exclure un déficit multiple en sulfatases (maladie d’Austin). L’identification des mutations chez le malade permet une détection fiable des hétérozygotes dans les familles.
Le diagnostic moléculaire
La recherche des mutations est possible lorsque le gène de l’enzyme est connu, notamment pour faciliter les études familiales et essayer d’établir des corrélations génotype-phénotype qui restent limitées par l’hétérogénéité des mutations. Toutefois quelques mutations plus fréquentes ont été décrites dans la MPS IIIA en fonction de l’origine géographique : mutation R245H en Hollande (58% des allèles), en Australie (41% des allèles) et en Allemagne (35% des allèles), mutation 1092delC en Espagne (46% des allèles), mutation R74C en Pologne (56% des allèles), mutation R456H en Australie (43% des allèles) et en Espagne (32% des allèles), mutation S66W en Italie (29% des allèles),…
Le diagnostic prénatal
Il est possible par la mesure de l’activité enzymatique correspondante réalisée soit dans une biopsie trophoblastique prélevée à la 8–9ème semaine de grossesse, soit plus tardivement dans les amniocytes cultivés après amniocentèse après la 14ème semaine de grossesse, même si l’étude des glycosaminoglycanes dans le surnageant du liquide amniotique peut donner une première indication. Le diagnostic est également possible par la recherche des mutations identifiées chez le patient index après avoir vérifié la ségrégation des allèles par l’étude de l’ADN des parents.
Traitement
Traitement symptomatique
Le caractère progressif de la maladie de Sanfilippo, comme des autres mucopolysaccharidoses, nécessite une évaluation régulière clinique et paraclinique pour adapter au mieux le traitement symptomatique et prévenir certaines complications.
Le traitement des troubles du comportement et du sommeil est difficile. Il fait appel aux antihistaminiques ou aux neuroleptiques sédatifs au début de la maladie, plus qu’aux benzodiazépines qui peuvent avoir un effet paradoxal. Le méthylphénidate ne paraît pas efficace pour traiter l’hyperkinésie de ces enfants. Les antidépresseurs tricycliques (Laroxyl°, Tofranil°, Anafranyl°) ont un bon effet sédatif en particulier nocturne, peut-être du fait de leur action antalgique sur d’éventuelles douleurs neuropathiques. La mélatonine semble donner d’assez bons résultats pour régulariser le sommeil et retrouver un meilleur rythme veille-sommeil.
L’épilepsie est habituellement peu active et son traitement facile à équilibrer. Il repose en première intention sur le valproate de sodium, ou sur la carbamazépine ou l’oxcarbazépine dont on peut également espérer un effet bénéfique sur le comportement et/ou d’éventuelles douleurs neuropathiques. Les troubles extrapyramidaux sont peu sensibles aux traitements spécifiques en dehors de l’arrêt des médications neuroleptiques. Le baclofène peut être utile pour diminuer la spasticité.
Le recours à une prophylaxie de l’endocardite bactérienne en cas de soins dentaires est systématique dès lors qu’il existe une valvulopathie. Les soins dentaires nécessitent souvent une anesthésie en raison des troubles du comportement. La kinésithérapie d’entretien articulaire, et surtout les appareillages orthopédiques (par exemple des attelles nocturnes pour prévenir l’aggravation d’un équin chez un enfant encore marchant) sont souvent mal acceptés ; le recours à un corset est parfois nécessaire pour stabiliser une déformation scoliotique. De même, en cas de surdité perceptive, la mise en place et la tolérance d’un appareillage auditif peuvent être difficiles, comme l’évaluation de son bénéfice.
Traitement spécifique
Il n’y a pas actuellement de traitement spécifique disponible pour les maladies de Sanfilippo humaines.
Dans cette maladie, la greffe de moelle osseuse n’a pas d’indication : elle a été tentée mais n’a aucun effet sur le cours neurologique de la maladie même si elle est réalisée au stade présymptomatique. De même la thérapeutique enzymatique substitutive se heurte à l’obstacle de la barrière hémato-méningée.
Des études de thérapie génique sur des modèles animaux se développent en particulier dans les maladies de Sanfilippo de types A et B.