

A chaud

Journée Mondiale Sanfilippo
16-02-2023Stimuler l’activité des microglies pour contrer la pathologie neuronale dans le syndrome de Sanfilippo type B
11-02-2023MPS III (Sanfilippo A,B,C et D)
Professionnel
Mise en garde : Les informations figurant dans cette fiche détaillée sont rédigées par un ou plusieurs experts. Elles sont destinées aux professionnels de la santé. Il est donc important d’analyser ces informations avec son médecin traitant, à la lumière de sa propre situation.
La maladie de Sanfilippo ou mucopolysaccharidose de type III (MPS III) est une maladie de surcharge lysosomale, du groupe des mucopolysaccharidoses, caractérisée par une dégradation intellectuelle sévère et rapide. Cette maladie est constituée de quatre sous-types cliniquement proches mais biochimiquement distincts.
La maladie est sous-diagnostiquée (dysmorphie généralement peu marquée) ; c’est la plus fréquente des mucopolysaccharidoses en Hollande et en Australie avec une prévalence respective de 1/53 0000 et 1/67 000. La fréquence des différents sous-types varie selon les pays : sous-type A plus fréquent en Angleterre, Hollande et Australie, et sous type B plus fréquent en Grèce et Portugal, les types IIIC et IIID étant beaucoup plus rares.
Source : Atlas Médical VML- Docteur Bénédicte Héron, Professeur Irène Maire - Docteur Roseline Froissart - Février 2007
Métabolisme - Pathogénie - Génétique
Les enzymes déficitaires sont :
- l’héparane-N-sulfatase ou héparane sulfamidase pour la MPS IIIA,
- l’alpha-N-acétylglucosaminidase pour la MPS IIIB,
- l’acétyl-CoA :alpha-glucosaminide-N-acétyltransférase pour la MPS IIIC,
- la N-acétylglucosamine-6-sulfatase pour la MPS IIID.
Ces quatre enzymes sont indispensables pour la dégradation de l’héparane sulfate. Le déficit enzymatique entraîne donc une accumulation d’héparane sulfate dans les lysosomes de l’organisme, notamment du cerveau et son excrétion anormale dans les urines où il peut être détecté.
Les gènes de trois de ces quatre enzymes ont été localisés et clonés (celui de la MPS IIIA en 17q25.3 ; celui de la MPS IIIB en 17q21.1 ; celui de la MPS IIID en 12q14). Celui de la MPS IIIC a été localisé récemment sur le chromosome 8 en 8p11-8 q11. La transmission des quatre maladies de Sanfilippo se fait sur un mode récessif autosomique.
Les héparane sulfates jouent en particulier un rôle de régulateur dans la migration cellulaire, la guidance axonale, la synaptogénèse et la plasticité structurale des cellules neuronales expliquant que leur présence excessive puisse induire des dysfonctionnements importants au niveau cérébral. En outre, des changements secondaires dans les fonctions métaboliques et cellulaires ont été mis en évidence dans des modèles de MPS III (augmentations secondaires d’autres hydrolases acides, augmentation des concentrations en gangliosides GM2 et GM3, phénomènes inflammatoires,…). La relation entre ces modifications et les désordres neurologiques n’est pas élucidée.
Presentation Clinique

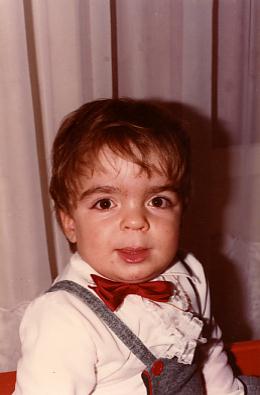
- Arnaud F. à 2 ans

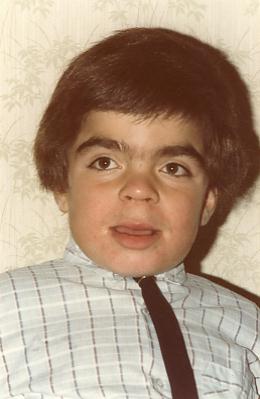
- Arnaud F. à 4 ans
Signes de début
Les premiers symptômes de cette maladie apparaissent habituellement entre 2 et 6 ans. Il s’agit principalement de troubles du comportement (hyperkinésie, agressivité) associés à des troubles du sommeil parfois importants, et à une stagnation des acquisitions (retard de langage, contrôle sphinctérien rarement acquis) et alors que les signes dysmorphiques sont modérés. Dans les formes précoces, des accès de terreur inexpliqués peuvent survenir dès les premiers mois de vie : le nourrisson hurle et déclenche un pseudo-Moro comme s’il tombait dans le vide ; l’enfant plus grand peut se mettre à pleurer en boucle comme s’il avait mal ou était inconfortable.
Ces enfants ont souvent une macrocéphalie, une taille normale voire une avance staturale au début, des traits un peu grossiers, un hirsutisme avec des cheveux épais et drus. Une hépatosplénomégalie modérée peut être retrouvée surtout chez les enfants jeunes. Des otites à répétition sont fréquemment notées depuis la première année de vie, avec une hypertrophie éventuelle des végétations adénoïdes et des amygdales, et une atteinte auditive mixte fréquente.
Les signes ostéo-articulaires sont plus discrets ou tardifs : un discret flessum des coudes est fréquent ; la raideur d’autres articulations (épaules, hanches, genoux) est plus tardive. Sur le plan radiologique, un éventuel aspect de rostre (sur les dernières vertèbres dorsales ou la première vertèbre lombaire) ou de dysostose multiple (habituellement minime ici) sont des éléments d’orientation aspécifiques vers une MPS.
La découverte d’un souffle cardiaque peut correspondre à une dysplasie valvulaire mitrale ou aortique, mais l’atteinte de la fonction cardiaque est tardive. Les épisodes récurrents de diarrhée motrice ont tendance à s’estomper avec l’âge. La survenue d’une puberté précoce est possible.
Evolution
Une déformation cypho-scoliotique du rachis ou une ostéochondrite des têtes fémorales complique plus rarement l’évolution orthopédique que dans les autres mucopolysaccharidoses.
L’évolution est marquée par une progression nette de l’atteinte neurologique à partir de la fin de la première décennie : régression puis perte des acquisitions psychomotrices (apraxie puis perte de la marche, de la station assise, de l’intérêt pour l’environnement et de la mastication-déglutition), atteinte pyramidale et/ou manifestations extrapyramidales (akinésie, rigidité plastique ou tremblements d’action ; parfois syndrome dystonique, ou dyskinétique souvent accentué ou favorisé par un traitement neuroleptique).
Une rétinopathie pigmentaire peut être observée. Une neuropathie périphérique, se manifestant par une amyotrophie et des troubles vasomoteurs à prédominance distale (en gants et en chaussettes) avec hyporéflexivité ostéotendineuse , peut être source de douleurs ; elle est de type axonal sensitivo-moteur à l’électromyogramme.
Une épilepsie survient souvent au cours de l’évolution, chez le grand enfant ou l’adolescent : les crises épileptiques sont le plus souvent généralisées et peu fréquentes, bien contrôlées par une monothérapie anti-épileptique.
L’imagerie cérébrale (scanner ou IRM) montre une atrophie corticale puis cortico-sous-corticale progressivement sévère tandis que se complètent la perte d’autonomie sociale, motrice, alimentaire et la perte de communication.
Les enfants atteints de cette maladie décèdent habituellement à la fin de la deuxième décennie dans un tableau de dégradation psychomotrice très sévère, associée à une tétraplégie spastique avec ses complications orthopédiques, des troubles de l’alimentation et du transit, des complications respiratoires qui nécessitent un traitement symptomatique aussi précoce et adapté que possible.
Les phénotypes cliniques des maladies de Sanfilippo A, B, C et D sont assez semblables mais sont hétérogènes y compris parfois dans une même famille. Le type A est plus souvent sévère avec un début plus précoce, et un décès vers la fin de la deuxième décade, mais des patients adultes ont été rapportés dont un cas de cardiomyopathie isolée et révélatrice chez un patient adulte. Les types B, C et D sont plus hétérogènes.
Diagnostic biologique
La mise en évidence de cellules de surcharge (cellules de Gasser,…) dans le sang ou la moelle peut constituer un élément d’orientation mais est peu spécifique.
Le diagnostic biochimique
![]() L’étude des glycosaminoglycanes (mucopolysaccharides) dans l’urine permet de mettre en évidence une présence anormale et isolée d’héparane sulfate confortant l’orientation diagnostique vers une maladie de Sanfilippo. Mais cette recherche peut être négative si l’on se contente d’une mesure quantitative ne séparant pas les différentes fractions excrétées de mucopolysaccharides, en particulier dans les formes modérées de l’adulte, car l’excrétion urinaire d’héparane sulfate diminue au-delà de l’adolescence. L’étude des glycosaminoglycanes urinaires s’est révélée complètement négative à la fois au plan quantitatif et qualitatif, dans une famille d’adultes atteints de MPS IIID à forme extrêmement modérée rapportée dans la littérature.
L’étude des glycosaminoglycanes (mucopolysaccharides) dans l’urine permet de mettre en évidence une présence anormale et isolée d’héparane sulfate confortant l’orientation diagnostique vers une maladie de Sanfilippo. Mais cette recherche peut être négative si l’on se contente d’une mesure quantitative ne séparant pas les différentes fractions excrétées de mucopolysaccharides, en particulier dans les formes modérées de l’adulte, car l’excrétion urinaire d’héparane sulfate diminue au-delà de l’adolescence. L’étude des glycosaminoglycanes urinaires s’est révélée complètement négative à la fois au plan quantitatif et qualitatif, dans une famille d’adultes atteints de MPS IIID à forme extrêmement modérée rapportée dans la littérature.
![]() La mise en évidence du déficit enzymatique se fait par la mesure de l’activité des différentes enzymes dans le sérum (MPS IIIB seulement) ou les leucocytes et les fibroblastes (pour les quatre sous-types). Pour les types IIIA et IIID, la mesure d’une autre sulfatase est indispensable pour exclure un déficit multiple en sulfatases (maladie d’Austin). L’identification des mutations chez le malade permet une détection fiable des hétérozygotes dans les familles.
La mise en évidence du déficit enzymatique se fait par la mesure de l’activité des différentes enzymes dans le sérum (MPS IIIB seulement) ou les leucocytes et les fibroblastes (pour les quatre sous-types). Pour les types IIIA et IIID, la mesure d’une autre sulfatase est indispensable pour exclure un déficit multiple en sulfatases (maladie d’Austin). L’identification des mutations chez le malade permet une détection fiable des hétérozygotes dans les familles.
Le diagnostic moléculaire
La recherche des mutations est possible lorsque le gène de l’enzyme est connu, notamment pour faciliter les études familiales et essayer d’établir des corrélations génotype-phénotype qui restent limitées par l’hétérogénéité des mutations. Toutefois quelques mutations plus fréquentes ont été décrites dans la MPS IIIA en fonction de l’origine géographique : mutation R245H en Hollande (58% des allèles), en Australie (41% des allèles) et en Allemagne (35% des allèles), mutation 1092delC en Espagne (46% des allèles), mutation R74C en Pologne (56% des allèles), mutation R456H en Australie (43% des allèles) et en Espagne (32% des allèles), mutation S66W en Italie (29% des allèles),…
Le diagnostic prénatal
Il est possible par la mesure de l’activité enzymatique correspondante réalisée soit dans une biopsie trophoblastique prélevée à la 8–9ème semaine de grossesse, soit plus tardivement dans les amniocytes cultivés après amniocentèse après la 14ème semaine de grossesse, même si l’étude des glycosaminoglycanes dans le surnageant du liquide amniotique peut donner une première indication. Le diagnostic est également possible par la recherche des mutations identifiées chez le patient index après avoir vérifié la ségrégation des allèles par l’étude de l’ADN des parents.
Traitement
Traitement symptomatique
Le caractère progressif de la maladie de Sanfilippo, comme des autres mucopolysaccharidoses, nécessite une évaluation régulière clinique et paraclinique pour adapter au mieux le traitement symptomatique et prévenir certaines complications.
Le traitement des troubles du comportement et du sommeil est difficile. Il fait appel aux antihistaminiques ou aux neuroleptiques sédatifs au début de la maladie, plus qu’aux benzodiazépines qui peuvent avoir un effet paradoxal. Le méthylphénidate ne paraît pas efficace pour traiter l’hyperkinésie de ces enfants. Les antidépresseurs tricycliques (Laroxyl°, Tofranil°, Anafranyl°) ont un bon effet sédatif en particulier nocturne, peut-être du fait de leur action antalgique sur d’éventuelles douleurs neuropathiques. La mélatonine semble donner d’assez bons résultats pour régulariser le sommeil et retrouver un meilleur rythme veille-sommeil.
L’épilepsie est habituellement peu active et son traitement facile à équilibrer. Il repose en première intention sur le valproate de sodium, ou sur la carbamazépine ou l’oxcarbazépine dont on peut également espérer un effet bénéfique sur le comportement et/ou d’éventuelles douleurs neuropathiques. Les troubles extrapyramidaux sont peu sensibles aux traitements spécifiques en dehors de l’arrêt des médications neuroleptiques. Le baclofène peut être utile pour diminuer la spasticité.
Le recours à une prophylaxie de l’endocardite bactérienne en cas de soins dentaires est systématique dès lors qu’il existe une valvulopathie. Les soins dentaires nécessitent souvent une anesthésie en raison des troubles du comportement. La kinésithérapie d’entretien articulaire, et surtout les appareillages orthopédiques (par exemple des attelles nocturnes pour prévenir l’aggravation d’un équin chez un enfant encore marchant) sont souvent mal acceptés ; le recours à un corset est parfois nécessaire pour stabiliser une déformation scoliotique. De même, en cas de surdité perceptive, la mise en place et la tolérance d’un appareillage auditif peuvent être difficiles, comme l’évaluation de son bénéfice.
Traitement spécifique
Il n’y a pas actuellement de traitement spécifique disponible pour les maladies de Sanfilippo humaines.
Dans cette maladie, la greffe de moelle osseuse n’a pas d’indication : elle a été tentée mais n’a aucun effet sur le cours neurologique de la maladie même si elle est réalisée au stade présymptomatique. De même la thérapeutique enzymatique substitutive se heurte à l’obstacle de la barrière hémato-méningée.
Des études de thérapie génique sur des modèles animaux se développent en particulier dans les maladies de Sanfilippo de types A et B.