

A chaud
MPS II avec atteinte neurologique : résultats de l’essai clinique d’un traitement enzymatique par voie intrathécale
23-02-2023
Webréunion - L’Anesthésie pour les MPS
28-11-2022MPS II (Hunter)
Professionnel
Mise en garde :
Les informations figurant dans la présente fiche ont été rédigées par un ou plusieurs experts, puis validées par un Comité Scientifique et Médical. Elle s’adresse par son contenu et sa description à des médecins. Une grande variabilité d’expression pouvant se manifester au sein d’une même maladie lysosomale, les informations présentées peuvent ne pas s’appliquer à un cas particulier. Il est donc important d’analyser ces informations avec son médecin traitant, à la lumière de sa propre situation.
Source : Atlas Médical VML - Docteur Nathalie GUFFON - Docteur Roseline FROISSART -Octobre 2006
La mucopolysaccharidose de type II, maladie de Hunter, est due au déficit en iduronate-2-sulfatase (IDS). La première description clinique de la maladie de Hunter date de 1917.
La mucopolysaccharidose de type II (MPS II), ou maladie de Hunter, est une maladie de surcharge lysosomale, du groupe des mucopolysaccharidoses. Sa prévalence est estimée entre 1/72 000 à 1/132 000 naissances masculines (extrêmement rare chez les filles).
Il existe un éventail de formes cliniques allant de formes sévères (les plus fréquentes), caractérisées par une régression psychomotrice précoce, à des formes atténuées.
L’enfant est normal à la naissance et ne présente pas de signes, qui n’apparaissant que progressivement.
Le tableau clinique des formes sévères associe hernies, dysmorphie faciale (macroglossie, bouche constamment entrouverte, traits épais), hépatosplénomégalie, limitations articulaires, syndrome du canal carpien, dysostose multiple, petite taille, troubles du comportement et dégradation psychomotrice aboutissant à une déficience intellectuelle, surdité, atteinte cardiaque et respiratoire, signes cutanés (aspect en peau d’orange au niveau des omoplates et des cuisses) ; par contre, les cornées sont classiquement claires.
Les formes atténuées sont caractérisées par une intelligence conservée, un syndrome dysmorphique et des dysostoses moins marqués et une survie prolongée.
La MPS II est due au déficit en iduronate 2-sulfatase (IDS) responsable de l’accumulation dans les lysosomes des différents tissus de dermatane-sulfate (DS) et d’héparane sulfate (HS). Le gène codant pour l’IDS est localisé en Xq28 et environ 320 anomalies géniques différentes ont été identifiées. Il s’agit de la seule mucopolysaccharidose transmise sur le mode récessif lié à l’X.
Seuls les garçons sont en principe touchés, mais environ douze cas de filles malades ont été décrits : dans la plupart des cas, une inactivation déséquilibrée de l’X entraînait une expression préférentielle de l’X muté.
L’excrétion urinaire accrue de DS et HS oriente le diagnostic biologique confirmé par la mise en évidence du déficit enzymatique en IDS dans le sérum, les leucocytes, ou les fibroblastes cultivés. La mesure d’une autre sulfatase est indispensable pour exclure un déficit multiple en sulfatases (maladie d’Austin). L’autre diagnostic différentiel est la mucopolysaccharidose de type I (MPS I - Hurler) pour les garçons.
Chez les femmes à risque, l’étude de l’activité enzymatique n’est pas un indicateur fiable de leur statut (conductrice ou non) du fait de la possibilité d’une inactivation déséquilibrée de l’X. Cette détermination peut être réalisée par étude moléculaire lorsque la mutation a été identifiée chez le malade. Le diagnostic prénatal n’est réalisé qu’en cas de foetus de sexe masculin, par étude de l’activité IDS ou par la recherche de la mutation identifiée chez le malade, après biopsie de trophoblaste ou amniocentèse.
En dehors des traitements symptomatiques, nécessitant le recours à une équipe multidisciplinaire, l’allogreffe de moelle osseuse n’est pas recommandée car elle n’empêche pas la détérioration intellectuelle.
Une thérapeutique enzymatique substitutive par perfusion d’enzyme recombinante (idursulfase) a obtenu l’AMM européenne (autorisation de mise sur le marché) en 2007 pour le traitement à long terme des patients. Les essais cliniques ont montré une amélioration de la marche et de l’atteinte respiratoire et des résultats significatifs sur la taille du foie ou de la rate et l’atteinte cardiaque. Il n’existe aucune donnée montrant une amélioration neurologique.
Pour les patients atteints de la forme la plus sévère, l’espérance de vie est très diminuée, le décès survenant généralement avant 20 ans par suite de complications cardio- respiratoires. Dans la forme modérée, les patients vivent jusqu’à l’âge adulte, parfois même jusqu’à plus de 60 ans pour les moins sévèrement atteints.
Métabolisme - Pathogénie - Génétique
L’iduronate-2-sulfatase hydrolyse spécifiquement le groupement sulfate en position 2 de l’acide iduronique présent dans le dermatane sulfate et l’héparane sulfate.
Le déficit en IDS est responsable de l’accumulation lysosomale progressive de ces deux glycosaminoglycanes entraînant des dysfonctionnements tissulaires et organiques. Compte tenu de leur large distribution, les manifestations de la mucopolysaccharidose de type II sont multisystémiques.
Le gène de l’IDS est situé sur la partie télomérique du bras long du chromosome X dans la région Xq28. Il comporte 9 exons et à une taille approximative de 24 Kb. Les études en biologie moléculaire ont permis de mettre en évidence dans 15 à 20% des cas une altération de grande taille (délétion totale ou partielle, réarrangements gène/pseudogène) et dans 80 à 85% des cas une altération de petite taille dont la plupart sont privées (plus de 300 mutations décrites).
Contrairement aux autres mucopolysaccharidoses, la transmission de la mucopolysaccharidose de type II se fait selon le mode récessif lié à l’X. De ce fait, seuls les garçons sont atteints. Les femmes conductrices transmettent leur X muté à la moitié de leurs enfants. A chaque grossesse, elles ont donc un risque de 50 % d’avoir un enfant atteint s’il s’agit d’un garçon et 50 % de donner naissance à une fille conductrice.
Une dizaine de cas de filles malades sont rapportés dans la littérature : dans la majorité des observations, ils sont expliqués par une inactivation déséquilibrée de l’X entraînant une expression préférentielle de l’X muté.
En l’absence de traitement simple et efficace de la maladie de Hunter, le dépistage des femmes conductrices est important afin de proposer un conseil génétique adapté. L’étude de l’activité IDS ne permet pas d’identifier de façon fiable les conductrices en raison de la possibilité d’une inactivation déséquilibrée de l’X. En conséquence, la détermination du statut de conductrice ou de non conductrice d’une femme à risque repose sur les études moléculaires quand la mutation du cas index est connue. L’incidence est estimée à environ 1/80 000 à 1/130 000 naissances masculines.
Présentation Clinique

- Cyphose dorsale lors de l’acquisition de la station assise
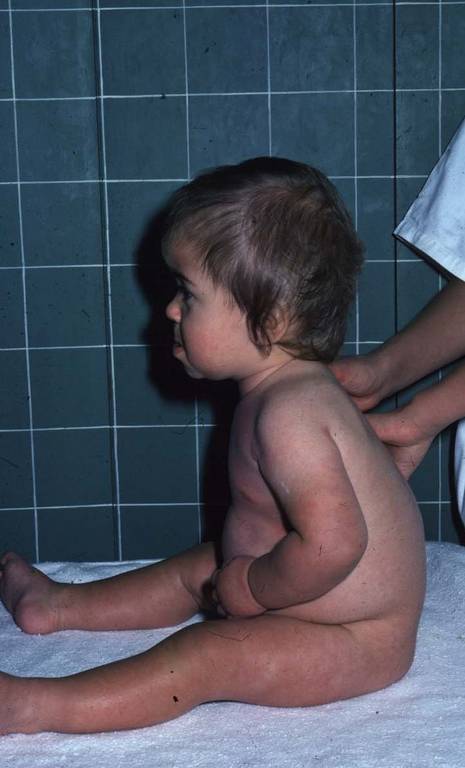
Cyphose dorsale lors de l’acquisition de la station assise
La présentation clinique de la mucopolysaccharidose de type II est très proche de celle de la mucopolysaccharidose de type I (maladie de Hurler). Là aussi, il existe un continuum de présentations cliniques allant de la forme la plus sévère à une forme atténuée avec une intelligence conservée.
Le diagnostic est souvent plus tardif en raison de signes dysmorphiques et osseux, souvent moins marqués que dans le type I. Le diagnostic n’est généralement posé qu’entre l’âge de 2 et 4 ans.
La mucopolysaccharidose de type II se différencie de la mucopolysaccharidose de type I essentiellement par les données suivantes :
- Syndrome dysmorphique et dysostoses moins marquées responsables du diagnostic tardif,
- Les cornées sont classiquement claires bien que chez certains adultes de discrets dépôts cornéens soient visibles lors de l’examen à la lampe à fente et qu’ils existent histologiquement,
- L’existence dans les formes sévères dès les 1ères années de vie de troubles du comportement importants : hyperactivité, obstination, agressivité, exubérance.
- La fréquence de signes cutanés réalisant un aspect de peau d’orange au niveau de l’omoplate et/ou des cuisses
L’ensemble des autres manifestations cliniques est décrit pour la mucopolysaccharidose de type I.
Diagnostic biologique
L’étude des glycosaminoglycanes (mucopolysaccharides) urinaires permet d’orienter le diagnostic. Les patients ont une excrétion accrue et qualitativement anormale (présence de dermatane sulfate et d’héparane sulfate). Ces anomalies ne permettent pas de distinguer la mucopolysaccharidose de type II de celle de type I. Les patients les plus jeunes et présentant une forme sévère ont une excrétion très élevée. Par contre, l’excrétion peut être peu augmentée chez les patients les plus modérés.
Le diagnostic de certitude repose sur la mise en évidence du déficit en IDS dans les leucocytes, dans le sérum ou dans les fibroblastes en culture. La valeur de l’activité enzymatique ne permet pas de distinguer les formes sévères des formes modérées.
La détermination d’une deuxième sulfatase est indispensable pour éliminer un déficit multiple en sulfatase ou maladie d’Austin.
L’étude du gène de l’IDS permet seulement dans certains cas de prédire la sévérité du phénotype (grande délétion et grand remaniement génique). L’étude génétique est indispensable pour la réalisation d’un conseil génétique adapté et le dépistage des conductrices.
Le diagnostic prénatal est proposé à toutes les femmes reconnues conductrices par l’étude de la généalogie ou les études moléculaires, aux conductrices potentielles dont le statut de conductrice n’a pas été déterminé, aux mères d’un enfant atteint même si elles ne sont pas reconnues conductrices, car on ne peut exclure l’existence d’une lignée de cellules germinales porteuses de l’X muté (mosaïcisme germinal).
Il est actuellement proposé une détermination du sexe fœtal par prélèvement sanguin effectué à la maman à 8 semaines d’aménorrhée. Si le fœtus est de sexe masculin, le diagnostic prénatal est possible par détermination de l’activité IDS ou par la recherche de la mutation identifiée chez le cas index, le plus souvent dans les villosités choriales prélevées vers 10-12 semaines d’aménorrhée (longueur crânio-caudale : 35-45 mm) avec une étude en direct qui permet un résultat rapide (inférieur à 1 semaine) et une interruption médicale de grossesse précoce si nécessaire.
Traitement
Les traitements symptomatiques sont importants et sont du même type que ceux de la mucopolysaccharidose de type I (maladie de Hurler).
Les traitements spécifiques : Les traitements visant à corriger le déficit enzymatique sont d’une part la transplantation de moelle osseuse et d’autre part le traitement enzymatique substitutif.
La transplantation de cellules souches hématopoïétique (moelle osseuse) : La maladie de Hunter n’est actuellement pas reconnue comme une indication de transplantation médullaire. Les patients ayant bénéficié d’une transplantation médullaire avec prise de greffe présentent une normalisation de leurs activités enzymatiques leucocytaires et de leurs excrétions urinaires des mucopolysaccharides au bout de 6 mois. La transplantation a un effet rapide sur l’infiltration des voies aériennes supérieures, sur l’hépatosplénomégalie, le morphotype, les lésions cardiaques sont stabilisées ou n’apparaissent pas, les amplitudes articulaires s’améliorent en poursuivant la kinésithérapie articulaire.
Le problème principal est le devenir cérébral des patients avec globalement trois groupes de réponses :
- Les patients qui de toutes façons n’auraient pas eu d’atteinte cérébrale tirent un bénéfice de l’amélioration de leur état viscéral et de la stabilisation de l’état cardio-respiratoire,
- Les patients atteints de la forme sévère présentant déjà des troubles du comportement avant la transplantation, présentent une régression psychomotrice plus lente et plus tardive avec perte de la marche à la puberté.
- Un groupe de patients intermédiaires pour lesquels il y a eu une amélioration des troubles du comportement, absence de perte des capacités motrices mais une impossibilité d’apprentissages scolaires, et/ou d’accès à une vie autonome.
Le traitement enzymatique substitutif :
Une thérapeutique enzymatique substitutive par perfusion d’enzyme recombinante (idursulfase) a obtenu l’AMM européenne (autorisation de mise sur le marché) en 2007 pour le traitement des patients. Les essais cliniques ont montré une amélioration de la marche et de l’atteinte respiratoire et des résultats significatifs sur la taille du foie ou de la rate et l’atteinte cardiaque. Il n’existe aucune donnée montrant une amélioration neurologique.
retour sur les essais cliniques ayant conduit à cette enzymotherapie :
L’iduronate-2-sulfatase humaine est produite par génie génétique dans des lignées de fibroblastes humains (laboratoire Shire, anciennement TKT).
Les essais de phase I/II ont été conduits aux Etats-Unis chez 12 patients atteints de MPS II. Dans ce premier essai humain, trois doses de traitement ont été testées (0.15 mg/Kg toutes les deux semaines, 0.5 mg/kg toutes les deux semaines et 1.5 mg/kg toutes des deux semaines). Un patient de chaque groupe recevait un placebo. L’étude a été conduite pendant six mois en double aveugle et se poursuit par une extension ouverte avec un traitement à 0.5 mg/kg toutes les deux semaines pour l’ensemble des patients. Ce premier essai a mis en évidence une efficacité clinique objectivée par la réduction de l’excrétion urinaire des mucopolysaccharides, des volumes spléniques et hépatiques et une augmentation de la distance parcourue en six minutes.
Le traitement est bien toléré avec des réactions associées à la perfusion plus importantes dans le groupe recevant 1.5 mg/kg toutes les deux semaines pour un gain clinique peu important.
De ce fait, la dose choisie pour poursuivre le traitement était de 0.5 mg/kg toutes les deux semaines.
Cette étude a été suivie d’un essai clinique de phase II/III randomisé en double aveugle contre placebo, multicentrique et multinational. Cet essai clinique a comporté une première phase d’un an dans laquelle ont été inclus 96 patients répartis en trois groupes : un groupe placebo (32 patients), un groupe recevant l’IDS recombinante à raison de 0.5 mg/kg toutes les deux semaines (32 patients) et un groupe recevant le traitement à raison de 0.5 mg/kg toutes les semaines (32 patients). L’âge d’inclusion allait d’environ 5 ans à 31 ans. Les patients devaient avoir une capacité vitale forcée inférieure à 80% et être capable de réaliser tous les tests demandés dans l’étude. Ils ne devaient avoir ni trachéotomie, ni avoir reçu une transplantation de cellules hématopoïétiques.
L’objectif primaire de cette étude est un score composite comportant le pourcentage de modification de la capacité vitale forcée et l’amélioration de la distance parcourue en 6 minutes.
L’ensemble des données de cette étude n’est pas encore disponible. Les trois groupes de patients étaient relativement homogènes. Les résultats préliminaires ont montré une réduction importante de l’excrétion urinaire des glycosaminoglycanes chez les patients traités quelle que soit la fréquence d’administration. Sur le plan des objectifs primaires, il existe une amélioration de la distance parcourue en 6 minutes dans les groupes traités alors qu’il n’y a pas de modification significative dans le groupe placebo. L’amélioration est plus importante dans le groupe recevant le traitement toutes les semaines (p = 0.0131) que dans le groupe traité tous les 15 jours (p = 0.07132). De même l’amélioration de la capacité vitale forcée est plus nette dans le groupe de patients recevant le traitement toutes les semaines, en particulier lorsque la modification est exprimée en valeur absolue, avec alors une amélioration statistiquement significative pour le groupe recevant le traitement toutes les semaines (p = 0.0011). La réduction de l’hépatosplénomégalie est rapide et significative dans les deux groupes.
Sur le plan des amplitudes articulaires, il n’y a pas de différence statistique entre les différents groupes pour l’évaluation globale. Néanmoins, il est à noter une amélioration dans la mobilité des épaules et des coudes dans le groupe recevant le traitement toutes les semaines par rapport au placebo.
Sur le plan de la sécurité du traitement, il a été noté des réactions liées à la perfusion chez 68 % des patients sans différence statistiquement significative entre les différents groupes. Ces réactions ont été facilement contrôlées par une prémédication et/ou un ralentissement de la vitesse de perfusion.
Environ 10 % des patients traités avec l’enzyme recombinante ont développé des anticorps.
Le dossier de demande d’autorisation de demande d’AMM européenne a été déposé en fin d’année 2005. La réponse positive de l’agence européenne du médicament est intervenu en 2006. En 2017, des expertises complémentaires sont en cours pour mesurer l’efficacité à moyen et long terme.