

Actualité de cette maladie
Chers Amis patients, parents et aidants, Nous vous invitons à consacrer 5/10 minutes pour (...)
MPS II (Hunter)
MPS II, une nouvelle stratégie thérapeutique
Dans la maladie de Hunter (MPSII), l’enzymothérapie substitutive, traitement spécifique apportant par voie intraveineuse l’enzyme IDS (Iduronate sulfatase) qui fait défaut aux malades, permet une diminution de la surcharge des GAG (glycosaminoglycanes) dans les cellules de différents organes du corps. Cependant, l’enzyme perfusée ne passe pas la barrière naturelle protectrice du cerveau (barrière hémato-encéphalique ou BHE) et ne peut donc pas agir sur les cellules du cerveau.
Lors du dernier congrès international des maladies lysosomales (WORLD – Orlando – 10/13 février 2020) auquel VML a pu assister, des chercheurs ont présenté les résultats d’une stratégie thérapeutique basée sur la conception d’une protéine de fusion pour apporter l’enzyme aux cellules du cerveau. L’idée est d’associer à l’enzyme IDS un fragment d’une autre protéine qui a la capacité de s’accrocher à un récepteur se trouvant à la surface de la BHE. Ce récepteur pourrait transporter la protéine de fusion à travers la barrière, permettant ainsi le passage de l’enzyme IDS jusqu’au cerveau.
Résultats d’études précliniques
Les chercheurs de la société de biotechnologie Denali Therapeutics, ont présenté leurs résultats précliniques (chez l’animal). Le nom de recherche de cette protéine de fusion est ETV:IDS (pour Enzyme Transport Vehicle : IDS), et son nom de développement industriel est DNL310).
Afin de prouver que cette approche permet l’apport de l’enzyme dans les types cellulaires majeurs du cerveau, les chercheurs ont perfusé chez des souris MPS II, l’ETV:IDS en intraveineux, une fois par semaine durant quatre semaines, à différentes concentrations.
Dans une première étape, ils ont constaté la présence de l’ETV:IDS ainsi qu’une diminution de la surcharge en GAG dans le cerveau et le liquide céphalo-rachidien et aussi dans le foie et la rate. Chez les souris MPS II perfusées par l’enzyme IDS classique, a présence de l’enzyme et les changements en surcharge des GAG dans le cerveau et le liquide céphalo-rachidien n’étaient pas retrouvés. La maladie MPS II induit également dans le cerveau une surcharge dite secondaire (par réaction en cascade) de lipides. Cette accumulation était également corrigée chez les souris traitées par ETV:IDS.
Les chercheurs ont ensuite isolé les neurones, les astrocytes et les cellules de la microglie, afin d’étudier la présence et l’action de l’ETV:IDS sur ces 3 types cellulaires majeurs du cerveau. L’ETV:IDS était bien retrouvée ainsi que la diminution en surcharge des GAG pour chacune. L’étude des cellules de la microglie a permis de montrer que l’ETV:IDS corrigeait la présence importante d’un marqueur de l’activation de ces cellules (CD68) ainsi que la variation dans l’expression de gènes liés à l’inflammation et à l’équilibre (homéostasie) du fonctionnement des lysosomes.
En outre, les chercheurs ont constaté dans le liquide céphalo-rachidien des souris traitées par ETV:IDS, une diminution de la quantité en NfL qui est un marqueur de la dégénérescence des neurones (plus spécifiquement de l’axone des neurones).
En conclusion, la protéine de fusion ETV:IDS permet de favoriser l’apport d’enzyme IDS au cerveau des souris MPS II. Cet apport se traduit par une diminution des différents substrats accumulés et une correction de marqueurs consécutifs à la maladie (dégénérescence des neurones, inflammation neuronale). Sur le temps alloué à leur présentation, les chercheurs n’ont malheureusement pas montré quel était l’impact de ces corrections cellulaires sur l’expression des symptômes cliniques.
Présentations orales par AG Henry et JC Ullman
Sur le site de déclaration des essais cliniques (Clinical Trial), Denali Therapeutics prévoit le lancement d’un premier essai clinique aux Etats-Unis courant 2020. Cet essai de phase I-II, a pour but principal d’évaluer la sécurité de ce traitement. Les patients MPS II devront être âgés de 2 à 18 ans et recevront une dose croissante de ETV:IDS (DNL 310) par perfusion intraveineuse toutes les semaines, sur une période de 24 semaines.
Résultats d’essais cliniques
Le laboratoire pharmaceutique japonais JCR Pharmaceuticalsa, quant à lui, montré les résultats de son essai clinique de phase II mené au Brésil (Présentation orale par R Giugliani), et les résultats intermédiaires de son essai clinique de phase II-III actuellement en cours au Japon (Présentation orale par T Okuyama).
Leur protéine de fusion, dénommée JR-141, avait préalablement fait l’objet d’un essai clinique de phase I-II au Japon, afin d’évaluer la sécurité et la dose de tolérance du traitement. Deux patients avaient reçu chaque semaine par voie intraveineuse, durant quatre semaines, une dose différente de JR-141 (0,01 mg/kg ; 0,1 mg/kg ; 1 mg/kg et 2 mg/kg). Ces doses ayant été bien tolérées, deux groupes de 6 patients avaient été constitués. Durant 4 semaines, JR-141 était perfusée chaque semaine à la dose de 1 mg/kg aux patients du premier groupe et à celle de 2 mg/kg aux patients du second groupe. L’étude concluait à une bonne tolérance du traitement à ces doses et sur cette période. En outre, après trois semaines de traitement, la concentration en héparane sulfate avait significativement diminué dans le liquide céphalo-rachidien (liquide « baignant » la moelle épinière et le cerveau).
L’essai clinique de phase II qui s’est déroulé au Brésil, avait pour but premier d’évaluer la sécurité du traitement sur une période plus longue (26 semaines) et à trois doses différentes (1 mg/kg ; 2 mg/kg et 4 mg/kg). Au total 20 malades ont été inclus, ayant une forme sévère ou modérée de la maladie, naïfs de tout traitement, ou en traitement depuis au moins 6 mois par enzymothérapie classique (impliquant alors un changement pour le traitement en évaluation).
Durant les 26 semaines, il a été estimé qu’aucun des effets indésirables sévères observés n’était lié au traitement. Les réactions à la perfusion ont été majoritairement constatées pour les patients du groupe recevant la plus forte dose (4 mg/kg/semaine). Ces réactions ont impliqué, pour cinq des sept patients du groupe, un arrêt provisoire de la perfusion et/ou la nécessité d’une médication complémentaire. Parmi les autres critères évalués, une diminution de la concentration en héparane sulfate dans le liquide céphalo-rachidien était observée à 26 semaines de traitement. Le groupe de patients recevant la plus faible dose avait une diminution moins importante (et pas chez tous) que celle constatée pour les deux autres groupes. Les cliniciens ont également évalué la différence entre le quotient de développement des enfants malades et celui d’enfants non malades du même âge. Sans traitement, l’évolution de la maladie accentue cette différence dans le temps. Dix enfants sur les douze évalués, ayant une forme sévère de la maladie, avaient une différence qui restait stable ou qui était améliorée à 26 semaines de traitement. Les quatre enfants évalués, ayant une forme modérée de la maladie, restaient stables ou s’amélioraient.
Par ailleurs, taille du foie et de la rate, concentration dans le sérum du dermatane sulfate diminuaient chez les patients initialement naïfs de traitement recevant les doses de JR-141 à 2 ou 4 mg/kg/semaine, et restaient stables pour l’ensemble des patients ayant préalablement été traités par l’enzymothérapie classique (qui agit au niveau périphérique).
La conclusion de cet essai est que le traitement est toléré jusqu’à une dose maximale de 4 mg/kg/semaine, la dose optimale étant de 2 mg/kg/semaine. L’administration en intraveineux de la protéine de fusion JR-141 permet d’obtenir une diminution de la concentration en héparane sulfate dans le liquide céphalo-rachidien, et différents éléments semblent indiquer une efficacité du traitement - à confirmer par un essai clinique international multicentrique de phase III, en cours de préparation pour les Etats-Unis, l’Europe et le Brésil.
L’essai clinique de phase II/III actuellement en cours au Japon a pour objectif premier d’évaluer l’effet du traitement sur la concentration en héparane sulfate dans le liquide céphalo-rachidien. D’autres objectifs dits secondaires sont prévus, pour évaluer l’effet du traitement sur certains marqueurs biologiques de la maladie et sur des éléments cliniques (endurance, déficience cognitive, trouble comportemental, amplitude articulaire…), ainsi que la tolérance au traitement. Au total, 28 patients participent à cette étude, dont 10 avaient déjà participé à l’essai de phase I/II au Japon (). Tous les patients reçoivent, une fois par semaine, une perfusion de JR-141 à la dose de 2 mg/kg. La durée totale prévue est de 52 semaines (un an). Les résultats intermédiaires présentés ont été obtenus après 26 semaines (6 mois) de traitement. A ce stade, l’ensemble des malades présente une diminution de la concentration en héparane sulfate dans le liquide céphalo-rachidien. Entre la fin du premier essai et le début du second, les 10 patients concernés ont durant un an repris le traitement par enzymothérapie classique. Lors de cette période, la concentration en héparane sulfate dans le liquide céphalo-rachidien avait à nouveau augmenté.
A 26 semaines, la fonction cognitive et les troubles comportementaux restent stables ou s’améliorent, le langage s’améliore chez la plupart. La concentration en héparane et dermatane sulfate ainsi que le volume du foie et de la rate restent stables chez les patients qui étaient traités par enzymothérapie, et diminuent chez les trois patients initialement naïfs de tout traitement.
Durant cette période, les effets indésirables sévères constatés (7) n’ont pas été liés au traitement.
En conclusion, les résultats intermédiaires sont positifs pour le critère premier de l’essai et le traitement semble dans l’ensemble bien toléré. L’effet thérapeutique du traitement observé à 26 semaines sur les symptômes neurologiques et les atteintes périphériques reste à confirmer par des résultats à plus long terme. Une demande d’autorisation de commercialisation du traitement au Japon est envisagée pour cette année.
Comprendre
La maladie de Hunter, ou mucopolysaccharidose de type II (MPS II), est une maladie de surcharge lysosomale du groupe des mucopolysaccharidoses. La mucopolysaccharidose de type II est une maladie évolutive, progressive, multisystémique. Elle est d’origine génétique et rare.
La première description clinique de la maladie de Hunter date de 1917.
Sa prévalence est estimée entre 1/72 000 à 1/132 000 naissances masculines (extrêmement rare chez les filles). Cela nous permet d’estimer le nombre de malades en France à un chiffre entre 110 et 130 patients, et environ 6 nouveaux cas par an.
Il s’agit d’une maladie génétique transmise sur le mode récessif lié au chromosome X. De ce fait seuls les garçons sont en principe touchés.
Les filles sont porteuses de la maladie mais le diagnostic pour elles est rare - mais quelques cas de filles malades ont été décrits : dans la plupart des cas, une inactivation déséquilibrée de l’X entraînait une expression préférentielle de l’X muté.
Comprendre l’action biochimique de la maladie.
Sur un plan biochimique, la maladie de Hunter (MPS II) est due à un déficit en enzyme "iduronate 2-sulfatase" (IDS). L’enzyme IDS permet la dégradation (hydrolyse) de certaines substances appelées glycosaminoglycanes (GAG) (notamment dermatane-sulfate (DS) et d’héparane sulfate (HS)). Les GAG sont des substances complexes produites par l’organisme et que l’on retrouve dans tous les types de tissu conjonctif qui soutient les organes et les tissus. Il compose également le cartilage des os pendant la croissance, des articulations et des valves cardiaques.
Le déficit de l’enzyme IDS est responsable d’une accumulation lysosomale des glycosaminoglycanes (GAG) dans la plupart des cellules. Cette accumulation va entrainer des dysfonctionnements tissulaires et organiques, notamment sur les voies respiratoires, le cœur, le foie, la rate, les os, les articulations et le système nerveux central, provoquant une maladie évolutive.
Le gène codant pour l’IDS à l’origine de cette maladie est localisé sur le chromosome X en Xq28 et environ 320 anomalies géniques différentes ont été identifiées.
Plusieurs formes cliniques et sévérités de la maladie
Il existe un éventail de formes cliniques allant de formes sévères (les plus fréquentes), caractérisées par une régression psychomotrice précoce aboutissant à une déficience intellectuelle et neurologique, à des formes atténuées caractérisées par une intelligence conservée, un syndrome dysmorphique et des dysostoses moins marqués.
Généralement les signes absents à la naissance de l’enfant apparaissent progressivement vers 2 - 3 ans. Chaque patient présente sa propre évolution de la maladie mais certains symptômes sont courants (voir onglet Symptome et Diagnostic). Des signes de l’ordre de l’hyperactivité, obstination, agressivité peuvent être observés.
Pour les patients atteints de la forme la plus sévère, l’espérance de vie est très diminuée, de l’ordre de l’adolescence. Dans la forme modérée, les patients atteignent l’âge adulte, parfois même jusqu’à plus de 60 ans pour les moins sévèrement atteints.
Symptôme - Diagnostic
 Généralement les signes absents à la naissance de l’enfant apparaissent progressivement vers 2 - 3 ans. Chaque patient présente sa propre évolution de la maladie mais parmi les symptômes les plus courants, notons : visage aux traits marqués avec élargissement du crâne, des raideurs articulaires, augmentation du volume du foie et de la rate, insuffisance cardiaque chronique, broncho-pneumopathie obstructive accompagnée d’apnées du sommeil, troubles de l’élocution et de l’audition, syndrome du canal carpien et dans les formes graves, atteinte du système nerveux central (SNC). Des signes de l’ordre de l’hyperactivité, obstination, agressivité peuvent être observés.
Généralement les signes absents à la naissance de l’enfant apparaissent progressivement vers 2 - 3 ans. Chaque patient présente sa propre évolution de la maladie mais parmi les symptômes les plus courants, notons : visage aux traits marqués avec élargissement du crâne, des raideurs articulaires, augmentation du volume du foie et de la rate, insuffisance cardiaque chronique, broncho-pneumopathie obstructive accompagnée d’apnées du sommeil, troubles de l’élocution et de l’audition, syndrome du canal carpien et dans les formes graves, atteinte du système nerveux central (SNC). Des signes de l’ordre de l’hyperactivité, obstination, agressivité peuvent être observés.
C’est sur l’observation de ces symptômes que le médecin va orienter le patient vers un diagnostic biologique de la maladie.
Le diagnostic biologique de la maladie de Hunter.
Le premier test se fait sur les excrétions urinaires pour mettre en évidence une dose accrue des GAG (DS et HS / voir onglet "comprendre"). Des valeurs anormales indiquent la présence éventuelle d’une maladie de type MPS.
La MPS II sera confirmée ensuite par la mise en évidence du déficit enzymatique en IDS dans le sérum, les leucocytes, ou les fibroblastes cultivés. La mesure d’une autre sulfatase est indispensable pour exclure un déficit multiple en sulfatases (maladie d’Austin, une autre maladie lysosomale). Un autre diagnostic va permettre de différencier la maladie de Hunter avec une autre mucopolysaccharidose, la MPS de type I (maladie de Hurler).
Ce diagnostic pourra être complété par une étude génétique du gène codant l’IDS. Cette étude génétique sera indispensable pour la réalisation d’un conseil génétique adapté (en cas de nouvelle grossesse) et le dépistage des femmes conductrices (enfants, parents, fratrie).
Dépistage de la maladie de Hunter
Un test génétique est possible. Il sera proposé aux femmes ayant déjà un enfant malade (ou famille à risque dû à des antécédents), permettant ainsi d’envisager une grossesse en toute sécurité. Le test peut aussi permettre de diagnostiquer précocement la fratrie d’un enfant malade.
>> Pour les couples ayant déjà eu un enfant atteint (ainsi que les femmes ayant été dépistées porteuses et les familles à risque dû à des antécédents) peuvent bénéficier du diagnostic prénatal. Il est actuellement réalisé par un test génétique.
>> Pour la fratrie, il est possible de dépister cette maladie chez les personnes à risque (sœurs du malade) avant qu’elle ne se déclare. Les tests génétiques permettent d’identifier dans la famille du malade les porteurs sains et les personnes atteintes de la maladie qui n’auraient pas encore développé les symptômes.
En cas de résultats positifs, ce dépistage et diagnostic précoces vont permettre une meilleure prise en charge mais en l’absence de traitement spécifique, ils n’assurent pas une guérison.
Traitements
En l’absence de traitement simple et efficace, aujourd’hui on ne guérit pas de la maladie de Hunter, mais la prise en charge a été grandement améliorée ces dernières années.
En France, les patients bénéficient désormais d’un traitement par thérapeutique enzymatique substitutive par perfusion d’enzyme recombinante (idursulfase).
Ce traitement a obtenu l’AMM européenne (autorisation de mise sur le marché) en 2007 pour le traitement à court terme des patients (des études complémentaires sont en cours pour mesurer sont efficacité à moyen et long terme). Les essais cliniques ont montré une amélioration de la marche et de l’atteinte respiratoire et des résultats significatifs sur la taille du foie ou de la rate et l’atteinte cardiaque. Il n’existe aucune donnée montrant une amélioration neurologique.
Les patients se verront aussi proposer de nombreux traitements symptomatiques pour répondre aux différentes complications. (canal carpien, hyperactivité ...), et/ou un accompagnement psychologique.
La greffe de moelle osseuse longtemps proposée n’est plus reconnue comme une indication dans la maladie de Hunter. (améliorations limitées, peu ou pas d’effet sur le plan neurologique, risques chirurgicaux importants ...)
VML, votre association
VML est une association POUR et PAR les patients, parents, familles touchés par les maladies lysosomales... Chacun pourra y trouver information, conseil, partage, accompagnement, mais pourra aussi apporter à son tour son envie d’agir contre la maladie et le handicap pour qu’ensemble fassions avancer le combat !
>> Retrouvez d’autres parents et patients au sein du Groupe VML- Hunter.
Si l’une des forces de VML est de fédérer l’ensemble des maladies lysosomales, les parents et patients concernés par la maladie de Hunter peuvent aussi se retrouver plus spécifiquement dans le "groupe patho VML- Hunter". Réunis autour d’un référent volontaire, les participants ont pour habitude de partager et d’échanger par mail leurs expériences, leurs informations, etc. Le groupe peut aussi être porteur d’un projet spécifique et son activité est liée à l’action des membres du groupe. Chacun peut être actif et intervenir à tout moment de l’année. Les membres se rencontrent généralement à l’occasion du week-end annuel de VML qui a lieu en mai, ou lors d’une journée spécifique Hunter / MPS.
>> Le livret génétique et transmission dans la maladie de Hunter
Un livret VML sur la maladie de Hunter est disponible sur demande à l’association (gratuit adhérent VML). Il regroupe tout ce qu’il faut savoir sur la maladie (symptômes, traitements, diagnostic, transmission...).
>> Des fiches d’information
Parce qu’un certain nombre de questions reviennent fréquemment sur le plan social, VML édite des fiches d’informations pour aider ses adhérents dans leur quotidien (sécurité sociale et complémentaire, la PCH en établissement, droits MDPH, passage enfant-adulte...).
>> Des professionnels à vos côtés
Les adhérents de l’association VML peuvent s’appuyer sur une équipe professionnelle pour obtenir des informations, des réponses à leurs questions, de l’accompagnement et du soutien dans leur démarche. Ce service est assuré par la responsable scientifique, la psychologue et l’assistante sociale de l’association VML.
>> Les actions et missions de l’association VML
Consultez un aperçu de nos actions au profit des adhérents de l’association
>> Accompagner l’après
Ce n’est pas dans l’ordre des choses que de voir partir son enfant, de jeunes adultes ou son conjoint. Dans ces moments-là, l’association VML reste à vos côtés et propose des temps de rencontres spécifiques.
Professionnel
Mise en garde :
Les informations figurant dans la présente fiche ont été rédigées par un ou plusieurs experts, puis validées par un Comité Scientifique et Médical. Elle s’adresse par son contenu et sa description à des médecins. Une grande variabilité d’expression pouvant se manifester au sein d’une même maladie lysosomale, les informations présentées peuvent ne pas s’appliquer à un cas particulier. Il est donc important d’analyser ces informations avec son médecin traitant, à la lumière de sa propre situation.
Source : Atlas Médical VML - Docteur Nathalie GUFFON - Docteur Roseline FROISSART -Octobre 2006
La mucopolysaccharidose de type II, maladie de Hunter, est due au déficit en iduronate-2-sulfatase (IDS). La première description clinique de la maladie de Hunter date de 1917.
La mucopolysaccharidose de type II (MPS II), ou maladie de Hunter, est une maladie de surcharge lysosomale, du groupe des mucopolysaccharidoses. Sa prévalence est estimée entre 1/72 000 à 1/132 000 naissances masculines (extrêmement rare chez les filles).
Il existe un éventail de formes cliniques allant de formes sévères (les plus fréquentes), caractérisées par une régression psychomotrice précoce, à des formes atténuées.
L’enfant est normal à la naissance et ne présente pas de signes, qui n’apparaissant que progressivement.
Le tableau clinique des formes sévères associe hernies, dysmorphie faciale (macroglossie, bouche constamment entrouverte, traits épais), hépatosplénomégalie, limitations articulaires, syndrome du canal carpien, dysostose multiple, petite taille, troubles du comportement et dégradation psychomotrice aboutissant à une déficience intellectuelle, surdité, atteinte cardiaque et respiratoire, signes cutanés (aspect en peau d’orange au niveau des omoplates et des cuisses) ; par contre, les cornées sont classiquement claires.
Les formes atténuées sont caractérisées par une intelligence conservée, un syndrome dysmorphique et des dysostoses moins marqués et une survie prolongée.
La MPS II est due au déficit en iduronate 2-sulfatase (IDS) responsable de l’accumulation dans les lysosomes des différents tissus de dermatane-sulfate (DS) et d’héparane sulfate (HS). Le gène codant pour l’IDS est localisé en Xq28 et environ 320 anomalies géniques différentes ont été identifiées. Il s’agit de la seule mucopolysaccharidose transmise sur le mode récessif lié à l’X.
Seuls les garçons sont en principe touchés, mais environ douze cas de filles malades ont été décrits : dans la plupart des cas, une inactivation déséquilibrée de l’X entraînait une expression préférentielle de l’X muté.
L’excrétion urinaire accrue de DS et HS oriente le diagnostic biologique confirmé par la mise en évidence du déficit enzymatique en IDS dans le sérum, les leucocytes, ou les fibroblastes cultivés. La mesure d’une autre sulfatase est indispensable pour exclure un déficit multiple en sulfatases (maladie d’Austin). L’autre diagnostic différentiel est la mucopolysaccharidose de type I (MPS I - Hurler) pour les garçons.
Chez les femmes à risque, l’étude de l’activité enzymatique n’est pas un indicateur fiable de leur statut (conductrice ou non) du fait de la possibilité d’une inactivation déséquilibrée de l’X. Cette détermination peut être réalisée par étude moléculaire lorsque la mutation a été identifiée chez le malade. Le diagnostic prénatal n’est réalisé qu’en cas de foetus de sexe masculin, par étude de l’activité IDS ou par la recherche de la mutation identifiée chez le malade, après biopsie de trophoblaste ou amniocentèse.
En dehors des traitements symptomatiques, nécessitant le recours à une équipe multidisciplinaire, l’allogreffe de moelle osseuse n’est pas recommandée car elle n’empêche pas la détérioration intellectuelle.
Une thérapeutique enzymatique substitutive par perfusion d’enzyme recombinante (idursulfase) a obtenu l’AMM européenne (autorisation de mise sur le marché) en 2007 pour le traitement à long terme des patients. Les essais cliniques ont montré une amélioration de la marche et de l’atteinte respiratoire et des résultats significatifs sur la taille du foie ou de la rate et l’atteinte cardiaque. Il n’existe aucune donnée montrant une amélioration neurologique.
Pour les patients atteints de la forme la plus sévère, l’espérance de vie est très diminuée, le décès survenant généralement avant 20 ans par suite de complications cardio- respiratoires. Dans la forme modérée, les patients vivent jusqu’à l’âge adulte, parfois même jusqu’à plus de 60 ans pour les moins sévèrement atteints.
Métabolisme - Pathogénie - Génétique
L’iduronate-2-sulfatase hydrolyse spécifiquement le groupement sulfate en position 2 de l’acide iduronique présent dans le dermatane sulfate et l’héparane sulfate.
Le déficit en IDS est responsable de l’accumulation lysosomale progressive de ces deux glycosaminoglycanes entraînant des dysfonctionnements tissulaires et organiques. Compte tenu de leur large distribution, les manifestations de la mucopolysaccharidose de type II sont multisystémiques.
Le gène de l’IDS est situé sur la partie télomérique du bras long du chromosome X dans la région Xq28. Il comporte 9 exons et à une taille approximative de 24 Kb. Les études en biologie moléculaire ont permis de mettre en évidence dans 15 à 20% des cas une altération de grande taille (délétion totale ou partielle, réarrangements gène/pseudogène) et dans 80 à 85% des cas une altération de petite taille dont la plupart sont privées (plus de 300 mutations décrites).
Contrairement aux autres mucopolysaccharidoses, la transmission de la mucopolysaccharidose de type II se fait selon le mode récessif lié à l’X. De ce fait, seuls les garçons sont atteints. Les femmes conductrices transmettent leur X muté à la moitié de leurs enfants. A chaque grossesse, elles ont donc un risque de 50 % d’avoir un enfant atteint s’il s’agit d’un garçon et 50 % de donner naissance à une fille conductrice.
Une dizaine de cas de filles malades sont rapportés dans la littérature : dans la majorité des observations, ils sont expliqués par une inactivation déséquilibrée de l’X entraînant une expression préférentielle de l’X muté.
En l’absence de traitement simple et efficace de la maladie de Hunter, le dépistage des femmes conductrices est important afin de proposer un conseil génétique adapté. L’étude de l’activité IDS ne permet pas d’identifier de façon fiable les conductrices en raison de la possibilité d’une inactivation déséquilibrée de l’X. En conséquence, la détermination du statut de conductrice ou de non conductrice d’une femme à risque repose sur les études moléculaires quand la mutation du cas index est connue. L’incidence est estimée à environ 1/80 000 à 1/130 000 naissances masculines.
Présentation Clinique

- Cyphose dorsale lors de l’acquisition de la station assise
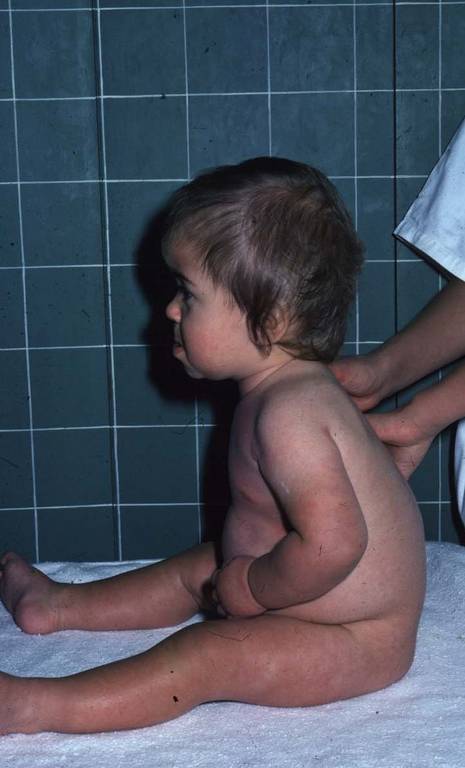
Cyphose dorsale lors de l’acquisition de la station assise
La présentation clinique de la mucopolysaccharidose de type II est très proche de celle de la mucopolysaccharidose de type I (maladie de Hurler). Là aussi, il existe un continuum de présentations cliniques allant de la forme la plus sévère à une forme atténuée avec une intelligence conservée.
Le diagnostic est souvent plus tardif en raison de signes dysmorphiques et osseux, souvent moins marqués que dans le type I. Le diagnostic n’est généralement posé qu’entre l’âge de 2 et 4 ans.
La mucopolysaccharidose de type II se différencie de la mucopolysaccharidose de type I essentiellement par les données suivantes :
- Syndrome dysmorphique et dysostoses moins marquées responsables du diagnostic tardif,
- Les cornées sont classiquement claires bien que chez certains adultes de discrets dépôts cornéens soient visibles lors de l’examen à la lampe à fente et qu’ils existent histologiquement,
- L’existence dans les formes sévères dès les 1ères années de vie de troubles du comportement importants : hyperactivité, obstination, agressivité, exubérance.
- La fréquence de signes cutanés réalisant un aspect de peau d’orange au niveau de l’omoplate et/ou des cuisses
L’ensemble des autres manifestations cliniques est décrit pour la mucopolysaccharidose de type I.
Diagnostic biologique
L’étude des glycosaminoglycanes (mucopolysaccharides) urinaires permet d’orienter le diagnostic. Les patients ont une excrétion accrue et qualitativement anormale (présence de dermatane sulfate et d’héparane sulfate). Ces anomalies ne permettent pas de distinguer la mucopolysaccharidose de type II de celle de type I. Les patients les plus jeunes et présentant une forme sévère ont une excrétion très élevée. Par contre, l’excrétion peut être peu augmentée chez les patients les plus modérés.
Le diagnostic de certitude repose sur la mise en évidence du déficit en IDS dans les leucocytes, dans le sérum ou dans les fibroblastes en culture. La valeur de l’activité enzymatique ne permet pas de distinguer les formes sévères des formes modérées.
La détermination d’une deuxième sulfatase est indispensable pour éliminer un déficit multiple en sulfatase ou maladie d’Austin.
L’étude du gène de l’IDS permet seulement dans certains cas de prédire la sévérité du phénotype (grande délétion et grand remaniement génique). L’étude génétique est indispensable pour la réalisation d’un conseil génétique adapté et le dépistage des conductrices.
Le diagnostic prénatal est proposé à toutes les femmes reconnues conductrices par l’étude de la généalogie ou les études moléculaires, aux conductrices potentielles dont le statut de conductrice n’a pas été déterminé, aux mères d’un enfant atteint même si elles ne sont pas reconnues conductrices, car on ne peut exclure l’existence d’une lignée de cellules germinales porteuses de l’X muté (mosaïcisme germinal).
Il est actuellement proposé une détermination du sexe fœtal par prélèvement sanguin effectué à la maman à 8 semaines d’aménorrhée. Si le fœtus est de sexe masculin, le diagnostic prénatal est possible par détermination de l’activité IDS ou par la recherche de la mutation identifiée chez le cas index, le plus souvent dans les villosités choriales prélevées vers 10-12 semaines d’aménorrhée (longueur crânio-caudale : 35-45 mm) avec une étude en direct qui permet un résultat rapide (inférieur à 1 semaine) et une interruption médicale de grossesse précoce si nécessaire.
Traitement
Les traitements symptomatiques sont importants et sont du même type que ceux de la mucopolysaccharidose de type I (maladie de Hurler).
Les traitements spécifiques : Les traitements visant à corriger le déficit enzymatique sont d’une part la transplantation de moelle osseuse et d’autre part le traitement enzymatique substitutif.
La transplantation de cellules souches hématopoïétique (moelle osseuse) : La maladie de Hunter n’est actuellement pas reconnue comme une indication de transplantation médullaire. Les patients ayant bénéficié d’une transplantation médullaire avec prise de greffe présentent une normalisation de leurs activités enzymatiques leucocytaires et de leurs excrétions urinaires des mucopolysaccharides au bout de 6 mois. La transplantation a un effet rapide sur l’infiltration des voies aériennes supérieures, sur l’hépatosplénomégalie, le morphotype, les lésions cardiaques sont stabilisées ou n’apparaissent pas, les amplitudes articulaires s’améliorent en poursuivant la kinésithérapie articulaire.
Le problème principal est le devenir cérébral des patients avec globalement trois groupes de réponses :
- Les patients qui de toutes façons n’auraient pas eu d’atteinte cérébrale tirent un bénéfice de l’amélioration de leur état viscéral et de la stabilisation de l’état cardio-respiratoire,
- Les patients atteints de la forme sévère présentant déjà des troubles du comportement avant la transplantation, présentent une régression psychomotrice plus lente et plus tardive avec perte de la marche à la puberté.
- Un groupe de patients intermédiaires pour lesquels il y a eu une amélioration des troubles du comportement, absence de perte des capacités motrices mais une impossibilité d’apprentissages scolaires, et/ou d’accès à une vie autonome.
Le traitement enzymatique substitutif :
Une thérapeutique enzymatique substitutive par perfusion d’enzyme recombinante (idursulfase) a obtenu l’AMM européenne (autorisation de mise sur le marché) en 2007 pour le traitement des patients. Les essais cliniques ont montré une amélioration de la marche et de l’atteinte respiratoire et des résultats significatifs sur la taille du foie ou de la rate et l’atteinte cardiaque. Il n’existe aucune donnée montrant une amélioration neurologique.
retour sur les essais cliniques ayant conduit à cette enzymotherapie :
L’iduronate-2-sulfatase humaine est produite par génie génétique dans des lignées de fibroblastes humains (laboratoire Shire, anciennement TKT).
Les essais de phase I/II ont été conduits aux Etats-Unis chez 12 patients atteints de MPS II. Dans ce premier essai humain, trois doses de traitement ont été testées (0.15 mg/Kg toutes les deux semaines, 0.5 mg/kg toutes les deux semaines et 1.5 mg/kg toutes des deux semaines). Un patient de chaque groupe recevait un placebo. L’étude a été conduite pendant six mois en double aveugle et se poursuit par une extension ouverte avec un traitement à 0.5 mg/kg toutes les deux semaines pour l’ensemble des patients. Ce premier essai a mis en évidence une efficacité clinique objectivée par la réduction de l’excrétion urinaire des mucopolysaccharides, des volumes spléniques et hépatiques et une augmentation de la distance parcourue en six minutes.
Le traitement est bien toléré avec des réactions associées à la perfusion plus importantes dans le groupe recevant 1.5 mg/kg toutes les deux semaines pour un gain clinique peu important.
De ce fait, la dose choisie pour poursuivre le traitement était de 0.5 mg/kg toutes les deux semaines.
Cette étude a été suivie d’un essai clinique de phase II/III randomisé en double aveugle contre placebo, multicentrique et multinational. Cet essai clinique a comporté une première phase d’un an dans laquelle ont été inclus 96 patients répartis en trois groupes : un groupe placebo (32 patients), un groupe recevant l’IDS recombinante à raison de 0.5 mg/kg toutes les deux semaines (32 patients) et un groupe recevant le traitement à raison de 0.5 mg/kg toutes les semaines (32 patients). L’âge d’inclusion allait d’environ 5 ans à 31 ans. Les patients devaient avoir une capacité vitale forcée inférieure à 80% et être capable de réaliser tous les tests demandés dans l’étude. Ils ne devaient avoir ni trachéotomie, ni avoir reçu une transplantation de cellules hématopoïétiques.
L’objectif primaire de cette étude est un score composite comportant le pourcentage de modification de la capacité vitale forcée et l’amélioration de la distance parcourue en 6 minutes.
L’ensemble des données de cette étude n’est pas encore disponible. Les trois groupes de patients étaient relativement homogènes. Les résultats préliminaires ont montré une réduction importante de l’excrétion urinaire des glycosaminoglycanes chez les patients traités quelle que soit la fréquence d’administration. Sur le plan des objectifs primaires, il existe une amélioration de la distance parcourue en 6 minutes dans les groupes traités alors qu’il n’y a pas de modification significative dans le groupe placebo. L’amélioration est plus importante dans le groupe recevant le traitement toutes les semaines (p = 0.0131) que dans le groupe traité tous les 15 jours (p = 0.07132). De même l’amélioration de la capacité vitale forcée est plus nette dans le groupe de patients recevant le traitement toutes les semaines, en particulier lorsque la modification est exprimée en valeur absolue, avec alors une amélioration statistiquement significative pour le groupe recevant le traitement toutes les semaines (p = 0.0011). La réduction de l’hépatosplénomégalie est rapide et significative dans les deux groupes.
Sur le plan des amplitudes articulaires, il n’y a pas de différence statistique entre les différents groupes pour l’évaluation globale. Néanmoins, il est à noter une amélioration dans la mobilité des épaules et des coudes dans le groupe recevant le traitement toutes les semaines par rapport au placebo.
Sur le plan de la sécurité du traitement, il a été noté des réactions liées à la perfusion chez 68 % des patients sans différence statistiquement significative entre les différents groupes. Ces réactions ont été facilement contrôlées par une prémédication et/ou un ralentissement de la vitesse de perfusion.
Environ 10 % des patients traités avec l’enzyme recombinante ont développé des anticorps.
Le dossier de demande d’autorisation de demande d’AMM européenne a été déposé en fin d’année 2005. La réponse positive de l’agence européenne du médicament est intervenu en 2006. En 2017, des expertises complémentaires sont en cours pour mesurer l’efficacité à moyen et long terme.