

Tay-Sachs
Généralité
Nous attirons votre attention sur le fait que la maladie évolue différemment chez chaque patient. Les informations d’ordre général contenues dans ces pages sont donc à prendre avec précautions et ne remplacent pas le lien avec vos médecins.} »
La maladie de Tay-Sachs, aussi appelé gangliosidose à GM2 variante B est une maladie génétique rare, neurodégénérative, de la famille des maladies lysosomales (cf qu’est ce qu’une maladie lysosomale). C’est en effet un mauvais fonctionnement du lysosome qui entraine une accumulation de "ganglioside GM2" dans les cellules.
C’est une maladie héréditaire à transmission autosomique récessive.(cf Comprendre la génétique et la transmission). Le gène responsable de la mutation à l’origine de la maladie est localisé sur le chromosome 15 (15q31).La prévalence de la maladie de Tay-Sachs est d’environ 1 cas pour 320 000 naissances.
La maladie de Tay-Sachs partage les signes cliniques avec la maladie de Sandhoff et sont différenciées depuis peu de temps.
Un peu d’histoire sur la découverte de la maladie de Tay-Sachs.
En 1881, l’ophtalmologiste britannique Warren Tay décrit la présence d’une tache rouge cerise sur la rétine d’un jeune enfant présentant un retard physique et mental. A la même période, le neurologue américain Bernard Sachs rapporte l’altération du développement cérébral chez des malades ayant un tableau clinique similaire. Cette maladie fut appelée maladie de Tay-Sachs.
Dans les années 60, on découvre que la substance principale qui s’accumule dans la maladie de Tay-Sachs est le ganglioside GM2. Cette substance est normalement dégradée par une enzyme, présente dans le lysosome, appelée hexosaminidase A (Hex A). Cette enzyme est un assemblage de 2 sous-unités : une sous-unité alpha et une sous-unité béta. Un défaut, d’origine génétique, dans la production de la sous-unité alpha entraine une défaillance de l’enzyme Hex A et donc l’accumulation du ganglioside GM2, essentiellement dans les cellules du cerveau.
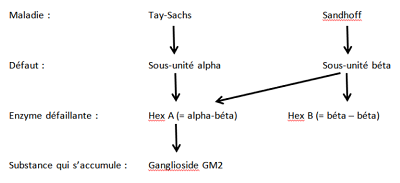 Il existe une autre hexosaminidase (hexosaminidase B = Hex B) qui est constituée par 2 sous-unités béta. Cette enzyme ne dégrade pas le ganglioside GM2 mais d’autres substances.
Il existe une autre hexosaminidase (hexosaminidase B = Hex B) qui est constituée par 2 sous-unités béta. Cette enzyme ne dégrade pas le ganglioside GM2 mais d’autres substances.
A la fin des années 60, Konrad Sandhoff, psychiatre et biochimiste, montre que chez certains malades les 2 hexosaminidases (A et B) sont défaillantes. Il s’agit d’un défaut dans la production de la sous-unité béta.
L’origine de la maladie étant différente pour ces malades, on a alors donné le nom de maladie de Sandhoff.
Il existe 3 formes de la maladie de Tay-Sachs selon l’âge de début.
La forme infantile (Tay-Sachs type 1) apparaît entre 3 et 6 mois. Le signe le plus précoce est l’apparition de sursauts inépuisables au bruit. Après 8 mois, l’enfant décline rapidement, un retard psychomoteur s’installant avec hypotonie et amaurose. La faiblesse musculaire augmente et aboutit à une paralysie. Les enfants présentent une mégaencéphalie, des crises d’épilepsie. Une tache rouge cerise au fond d’œil est présente mais non spécifique. La mort survient dans l’enfance dans un état de décérébration. Il existe un déficit très important voire une absence d’activité de l’hexosaminidase A dans les leucocytes ou les fibroblastes cultivés après biopsie de peau.
Le type juvénile (Tay-Sachs type 2) débute entre 2 et 6 ans. Il se traduit par une ataxie locomotrice, des troubles du comportement et une détérioration intellectuelle, aboutissant à une décérébration et au décès vers l’âge de 15 ans. La diminution de l’activité de l’hexosaminidase A est moins importante que dans la forme infantile.
La forme de l’adulte ou chronique (Tay-Sachs type 3) peut débuter vers l’âge de 10 ans mais le diagnostic n’est souvent fait qu’à l’âge adulte. Il existe deux tableaux cliniques. L’un ressemble à une maladie de Friedreich atypique avec une ataxie spino-cérébelleuse, sans signes cardiaques ni scoliose ou pieds creux. L’autre est celui d’une amyotrophie spinale juvénile ressemblant à un syndrome de Kugelberg-Welander. Il peut y avoir ou non une atteinte intellectuelle ou des troubles du comportement. Il existe un déficit en hexosaminidase A.
Deux variantes de la maladie ont été rapportées.
![]() La gangliosidose à GM2 variante B1 présente un tableau clinique identique aux formes juvénile et adulte des variantes B « classiques ». Le déficit en hexosaminidase A ne peut se détecter qu’avec un substrat artificiel particulier, différent de celui de la variante B.
La gangliosidose à GM2 variante B1 présente un tableau clinique identique aux formes juvénile et adulte des variantes B « classiques ». Le déficit en hexosaminidase A ne peut se détecter qu’avec un substrat artificiel particulier, différent de celui de la variante B.
![]() La gangliosidose à GM2 variante AB présente un tableau clinique d’une maladie de Tay-Sachs, mais l’activité de l’hexosaminidase A est normale. Il existe un déficit de l’activateur de l’enzyme nécessaire à l’hydrolyse du GM2. Le gène codant cette protéine est localisé sur le chromosome 5 (5q31).
La gangliosidose à GM2 variante AB présente un tableau clinique d’une maladie de Tay-Sachs, mais l’activité de l’hexosaminidase A est normale. Il existe un déficit de l’activateur de l’enzyme nécessaire à l’hydrolyse du GM2. Le gène codant cette protéine est localisé sur le chromosome 5 (5q31).
Auteurs : Prs N. Baumann et J.C. Turpin (avril 2006)
Delphine GENEVAZ - VML (2016)
Diagnostic et Traitement
Diagnostic
Certaines populations sont dites à risque (Juifs Ashkénazes), ou si un cas de la maladie a été connu dans la famille. Alors le dépistage des hétérozygotes et le diagnostic prénatal sont possibles et recommandés.
C’est un ensemble de signes qui pourront conduire à la suspicion d’une maladie de Tay-Sachs. La tache rouge cerise dans l’œil décrite par le docteur Tay en est un. Les Parents peuvent remarquer que l’enfant perd ou ne progresse pas dans l’apprentissage de se tenir et ramper, que l’enfant peut être effrayé par des bruits subits ou des mouvements (hyperreflexie). Parmi les signes les plus fréquents, notons :
- tache rouge cerise (Signe très fréquent)
- anomalie de l’audition/surdité (Signe très fréquent)
- cécité (Signe très fréquent)
- convulsions épilepsie (Signe très fréquent)
- diplégie/paraplégie/quadriplégie (Signe très fréquent)
- e.e.g. anormal (Signe très fréquent)
- hyperreflexie (Signe très fréquent)
- lipidose / sulfatidose (Signe très fréquent)
- régression psychique / démence (Signe très fréquent)
- retard mental sévère (Signe très fréquent)
- vision déficit modéré (Signe très fréquent)
- atrophie optique (Signe fréquent)
- hypertonie/rigidité/spasticité (Signe fréquent)
- hypotonie (Signe fréquent)
- myotonie (Signe fréquent)
Une prise de sang va permettre d’analyser la présence de la protéine HEXA dans le sang. Les patients présentant la Maladie de Tay-Sachs ne produisent pas ou pas assez de cette protéine (HEXA) de Hexosaminidase A. Ce test va permettre de confirmer la maladie de Tay-Sachs.
Traitement et recherche
Il n’existe pas encore de traitement spécifique à la maladie de Tay-Sachs. Les médecins peuvent tout de même proposer des traitements symptomatiques ;
◾ anti-épileptiques,
◾ lutte contre l’hypertonie,
◾ kinésithérapie motrice et respiratoire,
◾ amélioration de l’état nutritionnel,
◾ lutte contre la constipation.
La recherche de traitements
La thérapie génique permet de grands espoirs, mais les recherches sont encore longues. Un traitement qui vise à inhiber la synthèse des gangliosides (Miglustat) est à l’étude pour les formes lentement évolutives.
L’association VML s’engage au côté des chercheurs pour une meilleure compréhension de la maladie et la recherche d’un traitement.
VML, votre association
>> Retrouvez d’autres parents et patients au sein du Groupe VML Sandhoff / Tay-Sachs
Si l’une des forces de VML est de fédérer l’ensemble des maladies lysosomales dans un combat commun, les parents et patients concernés par les maladies de Sandhoff / Tay-Sachs peuvent aussi se retrouver plus spécifiquement dans le "groupe patho VML" qui leur est dédié.
>> Contacter le Groupe Sandhoff /Tay-Sachs
Réunis autour d’un ou plusieurs référents volontaires, les participants ont pour habitude de partager et d’échanger par mail leurs expériences, leurs informations, etc. Le groupe est ouvert aux parents qui vivent la maladie actuellement, ainsi qu’à ceux qui l’ont vécu et peuvent être des soutiens et conseils.
Le Groupe peut aussi être porteur d’un projet spécifique et son activité est liée à l’action des membres du groupe. Chacun peut être actif et intervenir à tout moment de l’année. Les membres se rencontrent généralement à l’occasion du week-end annuel de VML qui a lieu en mai, ou lors d’une journée spécifique Sandhoff / Tay-Sachs
>> Des professionnels à vos côtés
Les adhérents de l’association VML peuvent s’appuyer sur une équipe professionnelle pour obtenir des informations, des réponses à leurs questions, de l’accompagnement et du soutien dans leur démarche. Ce service est assuré par la responsable scientifique, la psychologue et l’assistante sociale de l’association VML, n’hésitez pas à les solliciter en contactant l’association.
>> Des fiches d’information
Parce qu’un certain nombre de questions reviennent fréquemment sur le plan social, VML édite des fiches d’informations pour aider ses adhérents dans leur quotidien.
>> Les actions et missions de l’association VML
Consultez un aperçu de nos actions au profit des adhérents de l’association
>> Accompagner l’après
Ce n’est pas dans l’ordre des choses que de voir partir son enfant, de jeunes adultes ou son conjoint. Dans ces moments-là, l’association VML reste à vos côtés et propose des temps de rencontres spécifiques.